
Reinheitsvalidierung von Medizinprodukten - Der Industrieverbund MediClean
24.11.2014 -
-
 FraunhoferIPA : Workshop am 03. Juli 2014 am Fraunhofer IPA in Stuttgart
FraunhoferIPA : Workshop am 03. Juli 2014 am Fraunhofer IPA in Stuttgart -
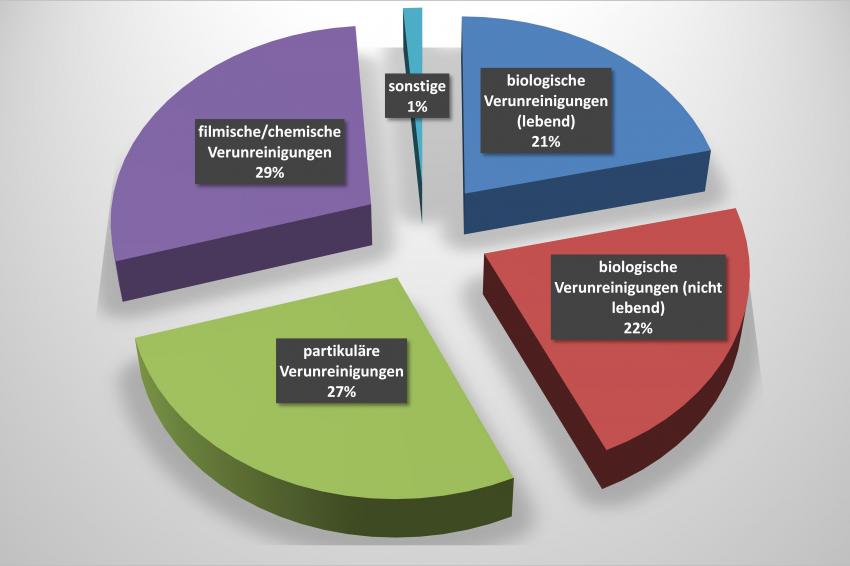 FraunhoferIPA: Relevanz unterschiedlicher Verunreinigungen im Zusammenhang mit Medizinprodukten aus Sicht der Medizintechnik-Experten [3]
FraunhoferIPA: Relevanz unterschiedlicher Verunreinigungen im Zusammenhang mit Medizinprodukten aus Sicht der Medizintechnik-Experten [3] -
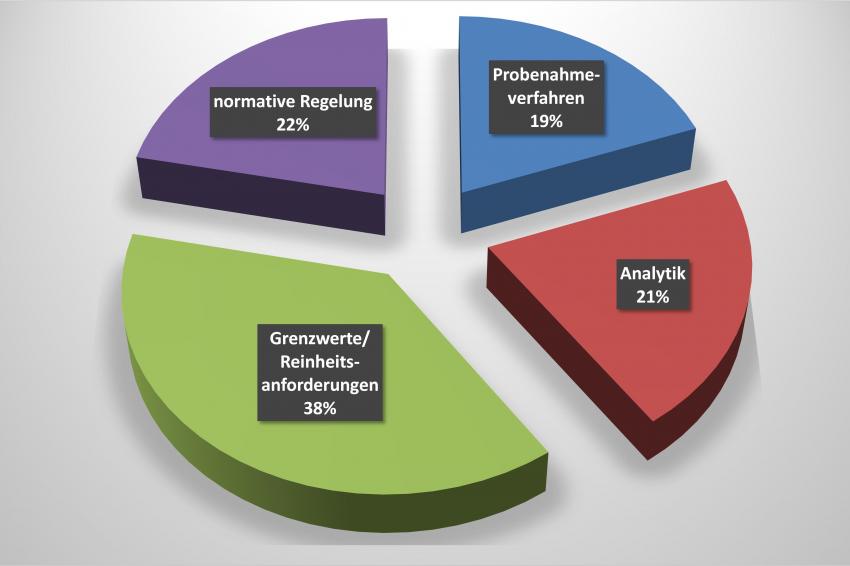 FraunhoferIPA : Zusammensetzung des Teilnehmerkreises
FraunhoferIPA : Zusammensetzung des Teilnehmerkreises -
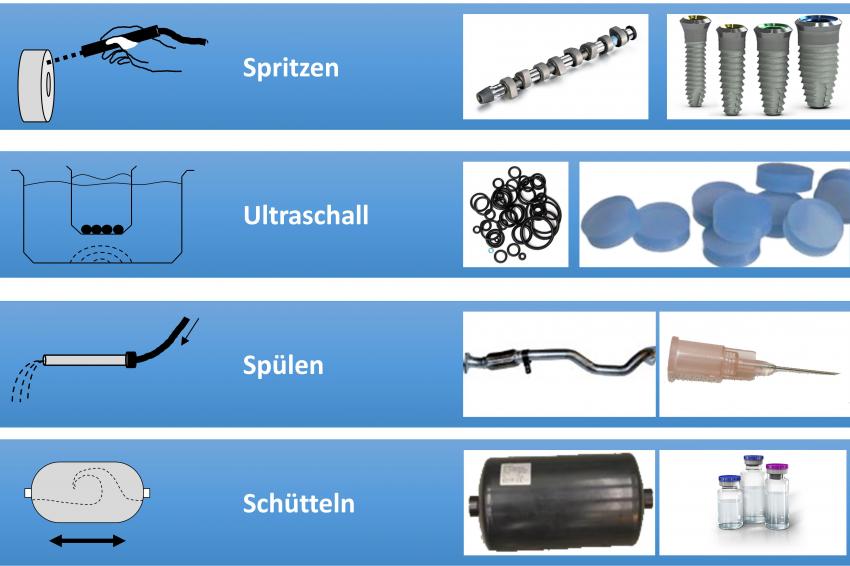 FraunhoferIPA: Übersicht der Probenahmeverfahren nach VDA Band 19
FraunhoferIPA: Übersicht der Probenahmeverfahren nach VDA Band 19 -
 FraunhoferIPA: Abklingmessung für ein Produkt zur Überprüfung der Wirksamkeit der Probenahme
FraunhoferIPA: Abklingmessung für ein Produkt zur Überprüfung der Wirksamkeit der Probenahme
In einer ständig älter werdenden Gesellschaft gewinnen medizintechnische Produkte zunehmend an Bedeutung. Höchste Qualitätsanforderungen sind bei ihrer Herstellung notwendig, um deren Langlebigkeit, Zuverlässigkeit und Sicherheit für den Anwender zu garantieren und damit den in der Regel geschwächten Patienten keinem unnötigen Risiko auszusetzen. In diesem Kontext rücken immer mehr Reinheitsaspekte in den Fokus.
Das normative Umfeld in der Medizintechnik regelt die Notwendigkeit von Reinheitsvalidierung, die Schwierigkeiten liegen aber in der Umsetzung: „Wie sauber ist sauber? Und wie analysiere ich Sauberkeit?", fragt Dr. Alexander Huwig, Senior Manager Research bei dem Hersteller von Zahnimplantaten Dentsply Implants Manufacturing in Mannheim.
Mit einem Workshop hat das Fraunhofer IPA einen Branchendialog initiiert, der über die Bildung des Industrieverbunds „MediClean" in ein branchenspezifisches und praxisnahes Regelwerks für die Reinheitsvalidierung von Medizinprodukten münden soll, das Einheitlichkeit und Vergleichbarkeit schafft.
Braucht die Medizintechnik neue Ansätze für die Reinheitsvalidierung?
Trotz des Einsatzes etablierter Reinraumtechnik können kritische Kontaminationen von Medizinprodukten nicht völlig ausgeschlossen werden. Rund ein Drittel aller Rückrufaktionen von Medizinprodukten durch die US Food and Drug Administration (FDA) zwischen 2001 und 2011 erfolgte aufgrund ungenügender Reinheit. Allein in Deutschland schätzen die Krankenkassen den ökonomischen Schaden durch Abstoßungsreaktionen des Körpers aufgrund unreiner Medizinprodukte wie Hüftimplantaten auf rund 7 Mrd. € jährlich. Der Eintrag von Verunreinigungen durch Personal, Equipment oder Prozessmedien kann damit die Sicherheit der Patienten ebenso wie die Zukunft eines Medizintechnik-Unternehmens gefährden.
Die Frage nach Reinheitsanforderungen, die maximalen Patientenschutz bieten und gleichzeitig in der Produktion einhaltbar sowie zuverlässig mit validierten Methoden überprüfbar sind, beschäftigt die Branche deshalb bereits seit längerer Zeit. Um den offenen Dialog darüber zu beginnen und den Handlungsbedarf abzuleiten, wurde am 3. Juli 2014 ein Workshop mit Experten aus Medizintechnik-Unternehmen und deren Zulieferern und Dienstleistern am Fraunhofer-Institut für Produktionstechnik und Automatisierung IPA in Stuttgart veranstaltet.
Ein wesentliches Ergebnis der Expertendiskussion war, dass die Reinheit von Medizinprodukten viel mehr als die rein biologischen Fragestellungen umfasst: Von den Teilnehmern wurden chemische bzw. filmische Kontaminationen (29 %) sowie Partikel (27 %) in diesem Zusammengang als gleichwertige, kritische Kontaminationen genannt.
In der Konsequenz müssen Medizinprodukte neben der Sterilität also auch auf filmische/chemische und partikuläre Reinheit untersucht werden. Im Zusammenhang mit diesen Reinheitsaspekten wurde von den Teilnehmern ein akuter Handlungsbedarf ermittelt: Abgesehen von der Sterilität, wurde das Fehlen von spezifischen Standards zur Überprüfung speziell der chemischen und partikulären Reinheit von Medizinprodukten festgestellt. Aus Sicht der Teilnehmer kann erst durch Schließen dieser Lücken der Aufwand, den jedes einzelne Medizintechnik-Unternehmen aktuell selbst zur Minimierung des Patientenrisikos betreiben muss, auf ein sinnvolles und praktikables Maß angepasst werden.
Aspekte im Rahmen der Reinigungsvalidierung
Akzeptanzkriterien
Für Medizinprodukte gibt es Reinheitsanforderungen, die in Form von Akzeptanzkriterien spezifiziert und damit bewertbar gemacht werden. Da es für Medizinprodukte aus Sicht partikulärer Kontaminationen zum Großteil noch keine eigens definierten Akzeptanzkriterien gibt, wird auf andere Branchen, z. B. die Pharmazie mit dem Europäischen Arzneibuch, zurückgegriffen. Die hier für Produkte wie Parenteralia geltenden Grenzwerte und Regelungen werden herangezogen, um sie auf Medizinprodukte zu übertragen. Spätestens bei der Überprüfung der Akzeptanzkriterien, die nach Arzneibuch entweder über Direktmessung der Flüssigkeit mit einem Partikelzähler oder manuelle, lichtmikroskopische Auswertung nach Abscheiden der Partikel auf eine Filtermembran erfolgen kann, ist eine direkte Übertragung auf das breite Spektrum unterschiedlichster Medizinprodukte nicht mehr möglich, da partikuläre Verunreinigungen nicht, wie im Falle der Parenteralia, bereits in einer Flüssigkeit vorliegen, sondern erst von der Oberfläche des Medizinprodukts extrahiert werden müssen. Mit der Existenz von Akzeptanzkriterien für ein Produkt ergibt sich aber zwangsläufig die Notwendigkeit, eine geeignete Vorgehensweise zu entwickeln, mit der die Akzeptanzkriterien zuverlässig überprüft werden können.
Probenahme als neuralgischer Punkt
Aufgrund der sehr breiten Produktpalette mit unterschiedlichsten geometrischen Merkmalen, ist die Anwendung eines universalen Probenahmeverfahrens für Medizinprodukte nicht möglich. Effektivität und Eignung des Probenahmeverfahrens, auch Extraktionsverfahren genannt, sind in diesem Zusammenhang ein neuralgischer Punkt, dem durch pauschale Anwendung eines Verfahrens nicht ausreichend Rechnung getragen wird.
Mit dieser Herausforderung sahen sich bereits andere reinheitskritische Branchen konfrontiert, die hierzu Lösungsansätze entwickelt haben. Die Lösung, die der Fragestellung der Medizintechnik am nächsten kommt, entstammt dabei der Automobilindustrie, die in ähnlicher Weise ein sehr breit gefächertes Bauteilspektrum aufweist. Der Nachweis technisch sauberer Komponenten erfolgt hier durch eine Sauberkeitsanalyse, die ein das gesamte Bauteil betrachtende Verfahren erforderlich macht, da sich der eine, vermeintlich funktionskritische Partikel auch in einer schwer zugänglichen Bauteilgeometrie befinden kann. Im Industrieverbund „Technische Sauberkeit (TecSa)" wurde unter Leitung des Fraunhofer IPA eine Lösung dieser Fragestellung mit den teilnehmenden Industrieunternehmen erarbeitet. Als Resultat wurde das Regelwerk „VDA Band 19 Prüfung der Technischen Sauberkeit - Partikelverunreinigung funktionsrelevanter Automobilteile" verfasst, in dem Ablauf und Varianten einer Sauberkeitsanalyse zur Bestimmung der partikulären Sauberkeit detailliert beschrieben werden. Eine Sauberkeitsanalyse lässt sich dabei im Wesentlichen in zwei Schritte unterteilen:
- Die Extraktion, bei der die Partikel mit Hilfe einer Flüssigkeit mit unterschiedlichen Verfahren (Spritzen, Ultraschall, Spülen, Schütteln) von der relevanten Produktoberfläche gewonnen werden und
- die eigentliche Analyse, die in den meisten Fällen mit einer Filtration beginnt, um die vom Bauteil extrahierten Partikel auf einen Analysefilter zu übertragen. Für die Analyse stehen ebenfalls unterschiedliche Verfahren zur Verfügung, wie Gravimetrie (Bestimmung des Rückstandsgewichts) und automatisierte mikroskopische Verfahren (Lichtmikroskopie sowie Rasterelektronenmikroskopie in Kombination mit energiedispersiver Röntgenspektroskopie EDX).
Bei der Erarbeitung einer geeigneten Methode zur Quantifizierung der partikulären Reinheit von Produkten müssen folgende Faktoren berücksichtigt werden:
- Blindwert:
Zur zuverlässigen Quantifizierung des Reinheitszustands von Produkten muss ausgeschlossen werden, dass die gewonnen Ergebnisse durch Personal, Equipment und Umgebung verfälscht werden. Da für Medizinprodukte bereits Partikel im Mikrometerbreich kritisch sein können, müssen in besonderer Weise reinheitstechnische Maßnahmen wie bspw. eine geeignete Reinraumumgebung berücksichtigt werden.
- Extraktionsverfahren:
Die Auswahl eines Extraktionsverfahrens erfolgt unter Berücksichtigung der Geometrie des Produkts ebenso wie unter Berücksichtigung der Verträglichkeit des Probenahmeverfahrens mit dem Produkt (Beschichtung, Material etc.). Die Wirksamkeit der Probenahme erfolgt über eine sogenannte Abklingmessung: Hierbei wird ein und dasselbe Bauteil wiederholt einer Extraktion unterzogen, um zu überprüfen, ob innerhalb von sechs aufeinanderfolgenden Extraktionsschritten das Abklingkriterium von 10 % erreicht wird. - Analyseverfahren: Die Auswahl eines zu verwendenden Analyseverfahrens kann von den in der Spezifikation formulierten Akzeptanzkriterien abgeleitet werden. Hier können nachzuweisende Partikelgröße sowie benötigter Aussagegehalt eine Rolle spielen.
Auf dem Weg zum Industrieverbund
Trotz der Brisanz der Reinheitsvalidierung von Medizinprodukten hat ein zielführender Dialog begonnen. Die zahlreichen Teilnehmer des IPA-Workshops im Juli sehen eindeutig Handlungsbedarf bei der Reinheitsvalidierung von Medizinprodukten. Ziel muss es ihrer Ansicht nach sein, ein verbindliches, branchenspezifisches und praxisgerechtes Regelwerk zu etablieren, das auch zukünftig international anerkannt ist.
Damit ein solches Regelwerk entstehen kann, waren die Hersteller in einer ungeahnten Offenheit bereit, ihre Erfahrungen und Expertise zur Verfügung zu stellen: „Wir möchten an der Findung der notwendigen Kriterien beteiligt sein", signalisierte ein Unternehmensvertreter während des Workshops die Bereitschaft, die Dinge selbst in die Hand zu nehmen. Das Fraunhofer IPA bot mit seinem Reinheitsforum die erste Gelegenheit, um sich zu treffen und notwendige Prozesse zu initiieren.
Durch die Einbindung möglichst vieler Spezialisten aus den Medizintechnik-Unternehmen und deren Zulieferer bei der Lösung der offenen Fragestellungen ist durch gemeinsame Konsensbildung gewährleistet, dass das Ergebnis auf eine breite Akzeptanz stößt. Bewährt hat sich in ähnlichen Fällen die Bildung eines Industrieverbunds. Ein gutes Beispiel liefert der Verbund „Technische Sauberkeit". Dort haben sich Automobilunternehmen und Zulieferer zusammengeschlossen, um gemeinsam die Beurteilung der partikulären Sauberkeit von Bauteilen und Komponenten in einem Regelwerk festzulegen (VDA Band 19).
Ähnlich diesem Vorbild wird das Fraunhofer IPA in den nächsten Wochen ein Treffen für Januar 2015 als Start für die Arbeit eines Industrieverbunds vorbereiten. Dieses offene Treffen wird allen Interessenten die Möglichkeit bieten, unverbindlich die Planung der Aktivitäten des Industrieverbunds für das Jahr 2015 kennenzulernen und gleichzeitig die spezifischen Inhalte mitzugestalten.
Bei Interesse an den detaillierten Ergebnissen und der Auswertung des Workshops sowie den nächsten Schritten auf dem Weg zum Verbund können Firmen sich per E-Mail an Guido Kreck wenden (guido.kreck@ipa.fraunhofer.de).
Auch können Sie gerne weiterhin Ihre Meinung zum aktuellen Stand der Reinheitsvalidierung von Medizinprodukten mitteilen, indem sie online an der Umfrage „Braucht die Medizintechnik neue Ansätze zur Reinheitsvalidierung?" teilnehmen: www.ipa.fraunhofer.de /medizintechnik
Alle Teilnehmer an der Umfrage erhalten bei Interesse eine detaillierte Auswertung.
Kontakt
Fraunhofer-Institut für Produktionstechnik und Automatisierung IPA
Nobelstr. 12
70569 Stuttgart
Deutschland
+49 711 970-1800
+49 711 970-1399









