The EXCiPACT Certification Scheme

There is a regulatory expectation that pharmaceutical companies will verify that the excipients produced by their suppliers are to an appropriate level of GMP. This has been achieved by auditing some suppliers, but today, the burden that generates on suppliers and their pharmaceutical company customers is both impractical and unsustainable.
Fortunately, the regulators support the verification of suitable GMP via independent, credible 3rd party certification schemes.
Excipient Regulation and Guidance
The pharmaceutical industry is increasingly using risk management principles to improve product quality and better protect patients. At the same time, regulators have called for more secure supply lines and defined quality measures for excipients. However, regulating pharmaceutical excipient quality is not easy as only a small percentage of excipients are made solely for pharmaceutical use. Whilst clearly defined GMP requirements already exist for APIs, defined GMP requirements for excipients were not formalized until recently.
In 2011, the EU’s Falsified Medicines Directive established that manufacturing authorization holders (MAH) must use a formalized risk assessment to ascertain the appropriate GMPs for ensuring excipient suitability. In 2015, the European Commission issued guidelines on the risk assessment for determination of the appropriate level of GMP for excipients. In 2016, EU drug product manufacturers (and those importing them into the EU) were required to implement risk assessments for the appropriate GMP for each excipient used. While regulations regarding GMP for APIs clearly define compliance needs, the responsibility for defining necessary GMPs for excipients in a specific drug product rests with the MAH holder.
Other countries are also developing formal requirements for excipient GMPs. The US Food Drug &Cosmetics Act specifies that drugs must be manufactured, processed, packed, and held in accordance with current good manufacturing practice (cGMP), or they are deemed to be adulterated. Under US law, a new pharmaceutical excipient, unlike an API, has no regulatory status unless it can be qualified through the approval mechanisms available for components used in finished drugs. The FDA assesses and permits use of excipients as part of a New Drug Application. In 2012, the FDA Safety and Innovation Act (FDASIA) Title VII became law, expanding the FDA’s authority to safeguard public health by, inter alia, enhancing the safety of the increasingly global drug supply chains. This requires drug manufacturers to include as part of a drug listing, the name, address, and unique facility identifiers of associated excipient manufacturers.
In Brazil, GMP requirements for excipients in locally sold drug products became law in 2016. In China, there are the new ‘bundling regulations’, consideration of local GMP regulations and, in 2015, an upgrade to the local pharmacopoeia. Other global initiatives affecting excipient quality include ICH Q3D for elemental impurities, ICH Q1B for stability requirements, QBD requirements, a Pharmacopoeial Discussion Group (PDG) monograph on harmonization, WHO initiatives on GMP and Good Trade and Distribution Practices.
Excipient Risk Assessment
Excipients come from many sources outside the pharmaceutical industry, including oil, plants, animals and mining. Over 1,400 excipients are used in drug products and the focus on risk assessments and transparency of supply chain integrity has become more prominent in regulations in recent years. However, unlike APIs, excipients are not made specifically for use in medicines. Rather they have a diversity of industrial uses from cosmetics to food which presents its own regulatory challenges. It would be impractical to directly regulate all excipient suppliers, and may even encourage some suppliers to leave the pharmaceutical sector. Therefore, the regulations require pharmaceutical drug manufacturers to conduct the risk assessment and ensure the excipient suppliers are of a suitable quality particularly as only the pharmaceutical manufacturer knows the intended use of the excipient.
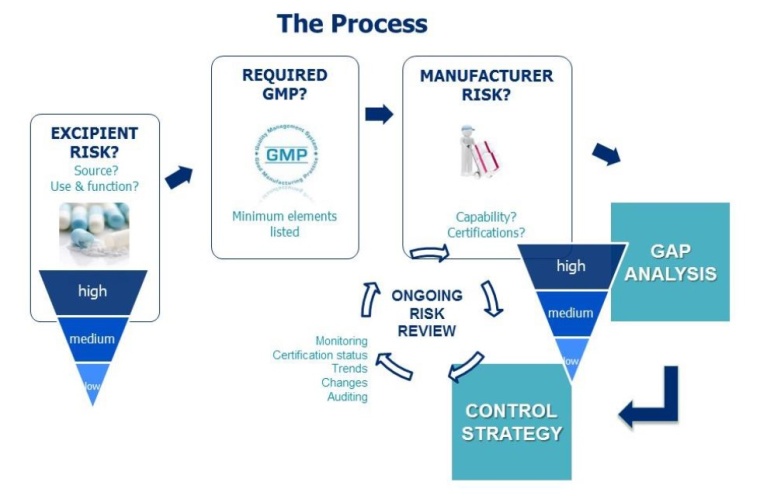
Patient safety is really the concern of the MAH and is not necessarily the direct focus of the excipient supplier. However, many excipient manufacturers do their own risk assessment and adopt suitable GMP to support business with their pharmaceutical customers. To help establish a consistent approach for the benefit of excipient suppliers and users, IPEC Europe and IPEC-Americas jointly published “The IPEC Risk Assessment Guide for Pharmaceutical Excipients, Part 1 – Risk Assessment for Excipient Manufacturers – First Version 2017.” This ‘How To’ document provides detailed guidance on a standardized approach to risk assessment as summarized in the process chart (see figure).
While all pharmaceutical manufacturers should now have the risk assessment in place to determine the appropriate GMP required for every excipient they use, questions remain on how supplier qualification should be implemented. Many prefer an audit of every supplier to provide evidence of GMP compliance. As the burden this would pose makes this untenable, supplier GMP certification schemes have become available. This approach is supported by regulators subject to certain conditions.
Excipient Certification
EC Guidelines permit the use of GMP certification held by the excipient manufacturer or supplier. While regulators have indicated that such approaches are acceptable, certain requirements must be met. These include that: the assessment must be against a standard that is suitable for excipient suppliers; the auditor must be demonstrably competent; and the certification body must have the quality management systems suitable for such a service, notably conformity to ISO 17021.
For example, the voluntary EXCiPACT GMP pharmaceutical excipient certification scheme meets these requirements and is being used as part of the risk assessment and supplier qualification process in a 3-year certification cycle that includes annual surveillance audits. Such certification assesses the quality management system of the excipient supplier together with the applied GMP, although may not meet the more demanding GMP requirements of, for example, parenteral drugs. It can also be used to certify the appropriate Good Distribution Practices (GDP) of the supply chain.
Such a certification scheme is one solution to meeting the new tougher regulatory requirements being faced by the pharmaceutical industry and its excipient suppliers. Certification is helping support supplier quality management while securing the supply chain and minimizing risk to patients.
References
- The European Parliament and The Council, Directive 2011/62/EU, Off. J. Eur. Union L174
https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2011_62/dir_2011_62_en.pdf
- Guidelines on the risk assessment for ascertaining the appropriate GMP for excipients of medicinal products, Off. J. Eur. Union C95
http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A52015XC0321%2802%29
- EudraLex Volume 4 EU Guidelines for GMP for Medicinal Products for Human and Veterinary Use, Part I Chapter 5 Production
https://ec.europa.eu/health/documents/eudralex/vol-4_en
- The IPEC Europe ‘How-To’ Document on EU Guidelines on Risk Assessment for Excipients
http://ipec-europe.org/UPLOADS/160318_IE_How-to_do_RAGuidelines_v1_2.pdf
- EPM Magazine September CPhI Issue 2016, ‘Pure Potential’ by Kevin McGlue, EXCiPACT President https://issuu.com/epmmagazineonline/docs/epm_16-6_mr_crop_rgb_link
- EXCiPACT Certification Standards for Pharmaceutical Excipient Suppliers: GMP and GDP, 2012
http://www.excipact.org/assets/Excipact-Standards.pdf
- The IPEC Risk Assessment for Pharmaceutical Excipients. Part 1 – Risk Assessment for Excipients Manufacturers http://ipec-europe.org/download.asp?fileToDL=IPEC_Risk_Assessment_Guide_FINAL_(May_8_2017).pdf
- US Food, Drug and Cosmetics Act 1938
https://www.fda.gov/aboutfda/whatwedo/history/productregulation/ucm132818.htm
- FDA Safety and Innovation Act, Title VII
Company
EXCiPACT





most read

Q1 2025 Chemical Industry: Diverging Trends
The first quarter of 2025 highlights a continued divergence between the European and US chemical industries.

Relocation of Chemicals Production Footprint in Full Swing
A new Horváth study based on interviews with CxOs of Europe’s top chemical corporations reveals: The majority of board members expects no or only weak growth for the current year.

Lead or Lag: Europe’s AI Materials Race
How AI and Robotics are reshaping the race for materials discovery.

20 Years of CHEManager International
Incredible but true: CHEManager International is celebrating its 20th anniversary!

ECA Foundation Aims to Become Largest Pharma Association for GMP/GDP Compliance
The ECA Foundation, one of the most important not-for-profit organizations for regulatory expertise in the pharmaceutical industry, aims to become the largest independent GMP/GDP organization in the world.