
Mikrobiologie in der Produktion
Mikrobiologische Qualifizierung und Validierung von Reinräumen in der Medizintechnik
-

-
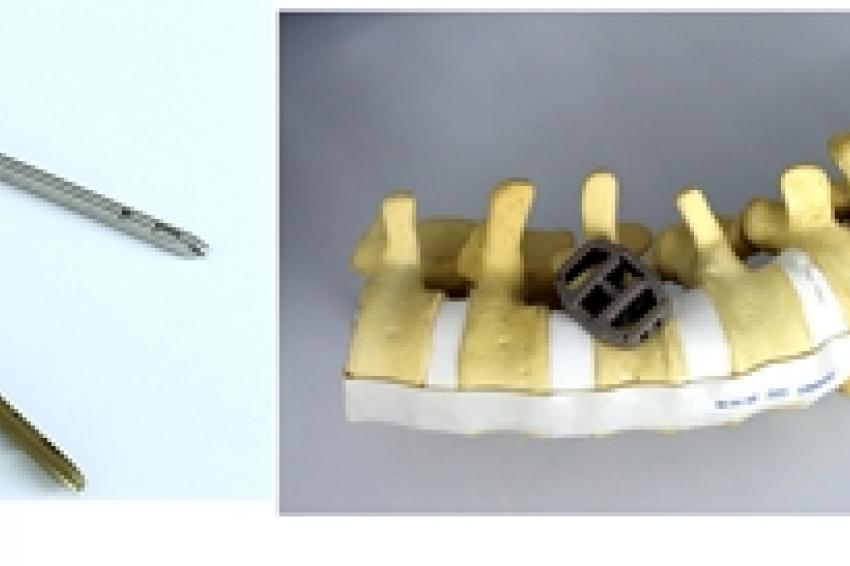 Das Pedikelschraubensystem VIPER 3D-MIS-Korrektur-Set für chirurgische Eingriffe an der Wirbelsäule.© DePuy Synthes Companies, Zuchwil
Das Pedikelschraubensystem VIPER 3D-MIS-Korrektur-Set für chirurgische Eingriffe an der Wirbelsäule.© DePuy Synthes Companies, Zuchwil -
 Operationsräume stellen kritische Zonen für eine mikrobielle Kontamination dar.© Wikipedia
Operationsräume stellen kritische Zonen für eine mikrobielle Kontamination dar.© Wikipedia -
 Die Herstellung von Medizinprodukten unterliegt einer strikten Regulation.© Gerresheimer AG
Die Herstellung von Medizinprodukten unterliegt einer strikten Regulation.© Gerresheimer AG -
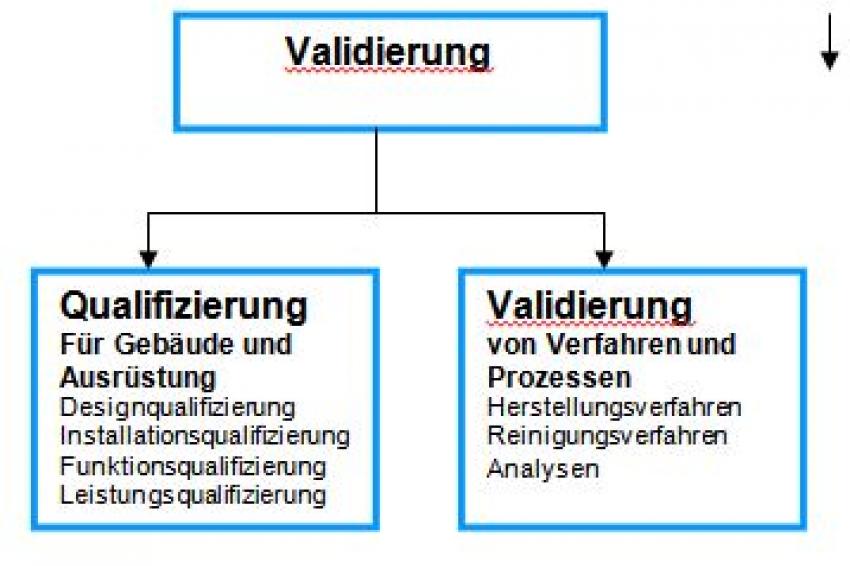 Validierung vs. Qualifizierung. Verfahren werden validiert, Räume und Geräte werden qualifiziert.
Validierung vs. Qualifizierung. Verfahren werden validiert, Räume und Geräte werden qualifiziert. -
 V-Modell nach GMP für Medizinprodukte. Für die Qualifizierung von neuen Anlagen ist dieses Grundschema verbindlich – mit den Spezifikationsschritten auf dem linken Teil, den entsprechenden Verifikationsschritten auf dem rechten.
V-Modell nach GMP für Medizinprodukte. Für die Qualifizierung von neuen Anlagen ist dieses Grundschema verbindlich – mit den Spezifikationsschritten auf dem linken Teil, den entsprechenden Verifikationsschritten auf dem rechten. -
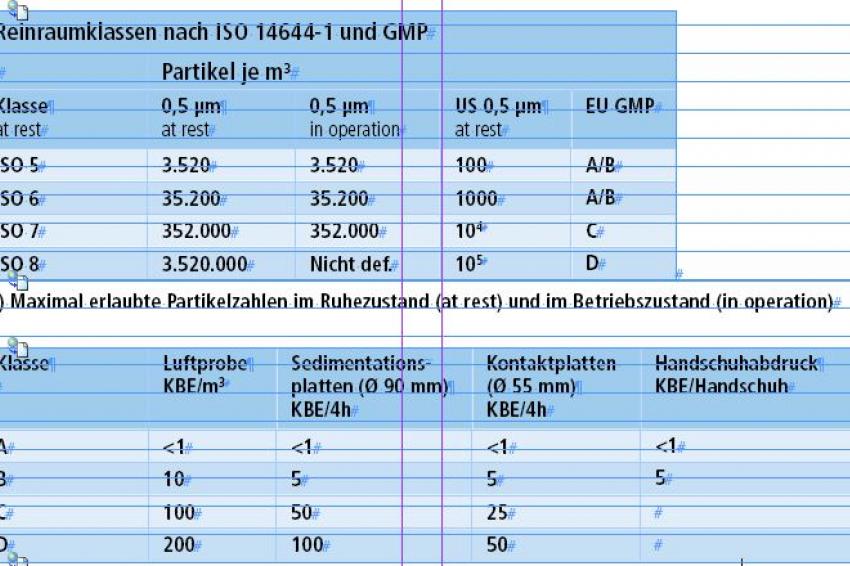 Reinraumklassen für die Pharmazie nach dem EU-GMP Leitfaden. In der Pharmazie und der Medizintechnik zählen Keime zu den Kontaminationsquellen. Die Norm zur Definition der Reinraumklassen wird hier durch den EU-GMP Leitfaden deklariert. (B) Isoklassen: Koloniebildende Einheiten (KBE): Empfohlene Grenzwerte für KBE.
Reinraumklassen für die Pharmazie nach dem EU-GMP Leitfaden. In der Pharmazie und der Medizintechnik zählen Keime zu den Kontaminationsquellen. Die Norm zur Definition der Reinraumklassen wird hier durch den EU-GMP Leitfaden deklariert. (B) Isoklassen: Koloniebildende Einheiten (KBE): Empfohlene Grenzwerte für KBE.
Die Herstellung von Medizinprodukten unterliegt besonderen Regularien. Denn Medizinprodukte müssen so ausgelegt und hergestellt sein, dass ihre Anwendung unter den vorgesehenen Bedingungen und zu den vorgesehenen Zwecken weder die Sicherheit des Patienten noch die Sicherheit und die Gesundheit der Anwender gefährdet.
Medizinische Produkte, z.B. chirurgische Instrumente, Geräte oder Implantate unterliegen strengeren Hygiene-Vorschriften als andere Reinraumprodukte, damit sie sicher am Patienten eingesetzt werden und keine Infektionen oder Entzündungen verursachen können. In der Medizinproduktebranche hat man es mit bewährten Verfahrensabläufen zu tun, die darauf beruhen, dass in solchen Produktionsketten ein Produkt partikel- und keimfrei hergestellt und steril bearbeitet wird. Denn Komplikationen, etwa Entzündungen des Patienten, können nicht nur durch Keime, sondern auch durch Partikel-Ablagerungen verursacht werden. Daher ist es wichtig, medizintechnische Produkte von vornherein kontaminationsfrei zu produzieren oder intensiv und rückstandfrei zu reinigen und möglichst wenig zu manipulieren. Denn wenn ein Produkt, das durch den Hersteller verkeimt oder mit Rückständen angeliefert wurde, im Krankenhaus sterilisiert wird, dann ist es nicht entkeimt, sondern es ist keimgetötet. Und auf der Oberfläche der Implantate oder Instrumente, mit denen am Menschen hantiert wird, haften dann immer noch Keim- und Partikel-Ablagerungen, die fiebrige Reaktionen beim Patienten erzeugen können. Daher muss die Keim- und Partikelbelastung so gering wie möglich gehalten werden. Dies hat Einfluss auf die Sterilisation. Im Hinblick auf die Produktsicherheit und Patientensicherheit sollte auch die Belastung mit Endo- und Produktionstoxinen minimal gehalten werden.
Validierung und Qualifizierung
Die Regulierung von Medizinprodukten ermöglicht den freien Warenverkehr in Europa und soll insbesondere sicherstellen, dass die Produkte grundlegende Anforderungen bezüglich Leistung und Sicherheit erfüllen. Sie stützt sich auf verschiedene Dokumente und Verordnungen, Staatsverträge, europäische Richtlinien, Auslegungen dieser Richtlinien, harmonisierte Normen und internationale Konsensdokumente und hat das Ziel, Patienten und Dritte zu schützen. Dabei unterliegt die Herstellung von Medizinprodukten weit schärferen Bedingungen als die technischer Produkte.
Bei der Validierung wird ein dokumentierter Beweis erbracht, dass ein Prozess oder ein System die vorher spezifizierten Anforderungen im Rahmen eines spezifischen Prüfverfahrens im praktischen Einsatz erfüllt. Bei mikrobiologischen Prüfverfahren steht die Spezifität und dass die Reinräume und Ausrüstungen hinsichtlich der Herstellungsprozesses auf ihre Eignung hin überprüft, bzw. qualifiziert, werden.
Die Qualifizierung ist ein mehrstufiger Prozess und umfasst die Planung, Konzeption und den Betrieb für Gebäude, Reinräume und Ausrüstungen. Die Details der Qualifizierung werden im EU-GMP-Leitfaden einer guten Herstellungspraxis definiert und regelmässig aktualisiert. Die europäischen Staaten sind verpflichtet, die Einhaltung der GMP-Regularien durchzusetzen.
Mikrobiologisch relevante Reinräume
Als Reinraum gilt ein nach DIN EN ISO 14644-1 und VDI 2083 betriebener Raum mit definierter Konzentration luftgetragener Partikel, der so konstruiert und verwendet wird, dass die Anzahl der in den Raum eingeschleppten bzw. im Raum entstehenden und abgelagerten Partikel kleinstmöglich ist und in dem andere reinheitsrelevante Parameter wie Temperatur, Feuchte und Druck nach Bedarf geregelt werden.
Heute gilt die ISO-Klassifizierung (ISO 5 bis ISO 8). Die ISO-Klassen entsprechen vier der bisherigen U.S.-Federal-Standard-209E-Klassen (siehe Tab. 1). Dabei entspricht im Betriebszustand Klasse A des EU-GMP-Leitfadens der ISO-Klasse 5/100, Klasse B der ISO-Klasse 7/10 000 und Klasse C der ISO-Klasse 8/100 000.
Für medizinische und pharmazeutische Anwendungen sind speziell neben der Partikelzahl auch die Keimzahlen von Bedeutung. Daher werden Medizinprodukte in der Regel im Reinraum oder in einem Containment hergestellt (Tab. 1 (B)). Bei der Planung und Implementation in der Medizinprodukte-Herstellung sind viel mehr Kriterien zu berücksichtigen als für rein technische Anwendungen. Die Planung muss die vielfältigen auf die Produktion einwirkenden Einflüsse wie Reinlufttechnik, Reinraumausrüstung, Prozessmedien, Prozesseinrichtungen, die Organisation und die Reinraummitarbeiter, berücksichtigen und sie in Einklang mit den strikten behördlichen Vorgaben bringen.
Regulatorische Anforderungen
Medizintechnikprodukte sind ähnlich wie Pharmaprodukte der GMP-Compliance unterworfen. Gemäss der ISO 13485 unterliegen sie der guten Herstellungspraxis und müssen die Qualitätsmanagement-Anforderungen entsprechend erfüllen.
Oberstes Ziel des Medizinproduktegesetzes ist es, die Gesundheit und den erforderlichen Schutz des Patienten zu gewährleisten. Es ist innerhalb der EU gültig und schreibt fest, was unter Medizinprodukten zu verstehen ist und wie mit ihnen verfahren wird. Für Medizinprodukte muss der Hersteller lückenlos nachweisen, dass bei der Herstellung das Risiko von Fehlern minimal ist und jeder die Qualität des Produkts beeinträchtigende Effekt vermieden wird.
Die ISO-Norm 13485 für Medizinprodukte-Qualitätsmanagementsysteme ist eine Art Durchführungsanleitung und schreibt fest, welche Qualitätskriterien bei der Herstellung von Medizinprodukten zu berücksichtigen sind. Diese Qualitätsanforderungen sind restriktiv formuliert.
Für die mikrobiologische Qualifizierung gelten der EU-GMP Guide - Annex 1 (2008, der EU-GMP Guide – Annex 15 (neue Version ab Okt. 2015)und das USP 1116: Microbiological Control and Monitoring of Aseptic Processing Environments.
Ziel: Kontaminationen vermeiden
Als Basis für die mikrobiologische Validierung dienen Normen wie die ISO 14644: Reinräume und zugehörige Reinraumbereiche, und Richtlinien wie die VDI2083 Reinraumtechnik.
Eine Qualifizierung gehört zum Validierungsprozess und umfasst Planung, Bau und Betrieb. Sie beinhaltet den Nachweis, dass der Betrieb gemäss den benutzerspezifischen Abläufen zertifiziert ist.
Dabei gibt es ein grundsätzliches Problem: Es ist mikrobiologisch nicht möglich zu beweisen, dass alle Keime durch einen Sterilitätsprozess inaktiviert worden sind. Sie können, weil unbekannt, nicht erkannt werden, oder weil die Anzuchtbedinungen nicht für einen Nachweis geeignet sind. Daher werden die meisten Hersteller eine Endsterilisation, entweder auf H2O2-Basis oder mit Strahlen, vornehmen.
In der Praxis beschreibt der Sterility Assurance Level (SAL), mit welcher Wahrscheinlichkeit eine einzelne Einheit nach dem Prozess nicht steril ist. Je besser der SAL Wert, desto effizienter ist der Sterilisations-Prozess.
QPM und URS
Der Ablauf einer Qualifizierung wird durch einen Qualifizierungsmasterplan (QPM) festgelegt. Er dient beim Audit (Inspektion) durch die Behörden als Grundlage für die Überprüfung. Ein Lastenheft (URS) beschreibt den technischen Leistungsumfang der gesamten Anlage. Die Behörden prüfen, ob die Anforderungen der URS bei der Planung umgesetzt wurden.
Die Qualifizierung wird in verschiedene Phasen eingeteilt:
Designphase
Die Designqualifizierung (DQ) gibt Aufschluss darüber, ob die Reinraumtechnische Produktionsanlage dem EU-Leitfaden entspricht. Die mikrobiologische Qualitätssicherung einer Anlage sollte bereits in dieser Designphase eingebunden sein!
Die Prüfung umfasst die Reinraumhülle, die Materialien bezüglich Reinigbarkeit und Desinfizierbarkeit, die Lüftung inklusive aller Einbauten sowie die Steuer- und Regelungstechnik.
Um Kontaminationsrisiken auszuschliessen müssen die einzelnen Einflussgrössen optimal aufeinander abgestimmt sein. Weil Verunreinigungen und Partikel vor allem über die Luft übertragen werden, müssen die Reinlufttechnischen Anlagen gemäss den Regularien ausgelegt sein.
Gibt es ungenügend durchströmte Bereiche im Produktionsraum? Liegt die Luftfeuchte oberhalb des Taupunktes? Eine Luftfeuchte über 60 % ist mikrobiologisch suboptimal. Eine Kondenswasserbildung durch Kühlelemente von Klimaanlagen kann zu starker Verkeimung führen. Diese gilt es zu vermeiden. Durch die Planung von Reinraum-Trocknern könnten schon in der Designphase Sicherheitsmargen in die Planung einfliessen. Bei eingeplanter Dekontamination des Reinraums durch Vernebeln ist eine korrekte Positionierung der Düsen einzuplanen.
In Bereichen mit hohem Personalbestand, wie etwa in Schleusen und Gängen, spielt der Luftwechsel eine grosse Rolle. Er muss ausreichend sein.
Bei der Prüfung der Reinigbarkeit müssen die Lösungsmittel hinsichtlich der Möglichkeit der sporiziden Desinfektion von Oberflächen mit starken Oxidationsmitteln, starken Säuren und Laugen hinterfragt werden.
Die DQ umfasst auch eine Risikoanalyse, in der die Produktionsräume auf Schwachstellen hin überprüft werden. Diese gründliche Prüfung der Planungsunterlagen verhindert, dass Planungsmängel erst in den Realisierungsphasen erkannt werden.
Installationsprüfung
Mit der Installationsqualifizierung (IQ) wird sichergestellt und dokumentiert, dass die Anlage so installiert wurde, wie es die normativen und benutzerspezifischen Anforderungen verlangen. Alle Herstellungsschritte müssen lückenlos dokumentiert werden, darunter z. B. auch die Qualität der Rohstoffe und die Art des Verpackungsmaterials. Gerade auch die Grenzwerte für eine mikrobiologische Kontamination mit keimbildenden Einheiten (KBE) sind strikt einzuhalten (s. Tab. 1). Diese Einhaltung muss nachgewiesen werden können. Viele Hersteller dokumentieren sie neben Eigenkontrollen zusätzlich durch Fremdkontrollen.
As built
Die As-built-Dokumentation stellt eine Gesamtdokumentation der Produktionsanlage dar, die die Anlage zum Zeitpunkt ihrer Abnahme (as built) vollständig gemäß vertraglicher Vereinbarung beschreibt. Sie besteht aus den Anlage- und Prüfdokumentationen zum Abnahmezeitpunkt.
Die Bestimmung der Ausgangskeimlast von Oberflächen ist nur in A und B Zonen sinnvoll. In dieser Phase kann die Bestimmung der Hauskeimflora sinnvoll sein.
Funktionsqualifizierung
Die Funktionsqualifizierung (OQ) soll dokumentieren, dass die Anlage und ihre Bestandteile im Rahmen der vorgesehenen Betriebsbereiche gemäß den Erwartungen funktionieren. Zu diesem Zweck sollten im Zuge des OQ-Plans Prüflisten für die Funktionsprüfung erstellt werden. Prüfpunkte sind zum Beispiel Funktions- und Sicherheitsprüfungen der Lüftungsanlage und des Reinraums sowie messtechnische Funktionsprüfungen im Zustand „at rest“.
Für die messtechnischen Prüfungen definiert der EU-GMP-Leitfaden die zwei Zustände „at rest“ (Ruhezustand) und „in operation“ (Betriebszustand). Der Zustand „at rest“ umschreibt die Situation, in der die Produktionsanlage installiert und ohne Anwesenheit des Bedienpersonals in Betrieb ist. Sie bietet den Nachweis, dass die Technik und das Design geeignet sind, die zonenspezifischen Anforderungen an die Luftkeimzahl zu erfüllen. Zudem liefert sie den Nachweis, dass Reinigungs- und Desinfektionsmittel in Kombination mit den Reinigungs- und Desinfektionsverfahren geeignet sind, die Oberflächen des Raums und der Einrichtung in den hygienisch sauberen Zustand zu überführen.
Der Zustand „in operation“ bedeutet dagegen, dass die Zonen-spezifischen Anforderungen an die Luft- und Oberflächenkeimzahl auch bei laufendem Betrieb mit Personal inklusive Materialfluss langfristig eingehalten werden. Dafür muss das Personal auf seine Aufgaben vorbereitet werden. Denn die mit Abstand grösste Quelle für mikrobiologische Kontaminationen ist der Mensch. Grosser Wert wird daher auf die Bekleidungsvorschriften und das Arbeitsverhalten im Reinraum gelegt.
Der mikrobiologische Validierungsplan
Mit einer Reinigungsvalidierung überprüft man ein genehmigtes Reinigungsverfahren, das für Anlagen oder Ausrüstungsteile in der Medizintechnik konzipiert wurde. Die Validierung dokumentiert den Nachweis, dass dieses Reinigungsverfahren geeignet ist, um eine anschliessende Herstellung von Medizintechnikprodukten durchzuführen. Der mikrobiologische Validierungsplan beinhaltet die Ziele, beschreibt den Umfang der Reinigungstätigkeiten, die Methodik sowie die Verantwortlichkeiten. Er beschreibt den Zweck der Validierung, die Anzahl der Validierungsläufe, die Prüfpositionen, die Prüfmethode (Luftkeimsammler, Abklatsch), mikrobiologische, zonenspezifische Anforderungen, die genaue Beschreibung des Ablaufs, vor allem „in operation“. Massgebend ist dabei die Dokumentation der Rohdaten, z. B. die des Personals im Reinraum, die durchgeführten Arbeiten, das Vorgehen bei Abweichungen, etc.
Der abschliessende mikrobiologische Validierungsbericht umfasst die Besonderheiten bei der Durchführung, die Ergebnisse und Abweichungen, eine Risiko-Analyse von Worst-Case-Szenarien, die in das Routineprogramm übernommen werden müssen sowie eine Festlegung, ob und wann eine Requalifizierung notwendig ist. Der Validierungsbericht muss vom Management her genehmigt werden.
Qualifizierungsabschlussbericht
Der Qualifizierungsabschlussbericht beinhaltet einen Dokumentennachweis mit allen während der Qualifizierung erstellten Dokumenten. Er kennzeichnet den Qualifizierungsstatuts der Produktionsanlage eindeutig. Er sollte eine zeitliche Vorgabe beinhalten, wann eine periodisch wiederkehrende Requalifizierung erforderlich wird. Diese Vorgaben sollten sich auf die Anlage beziehen. In vielen Fällen werden Reinräume alle drei bis fünf Jahre requalifiziert. Weitere Gründe für eine Requalifizierung sind Änderungen der Lüftungstechnik, Designänderungen, die Auswirkungen auf Luftführung, Material- und Personalfluss haben, z.B. Einführen grosser Maschinen, eine Änderung des Reinigungs- und Desinfektionsverfahrens. Keine periodische Requalifizierung ist notwendig, wenn ein regelmässiges Umgebungsmonitoring durchgeführt wird.
Sind schliesslich alle Systeme korrekt installiert und ist der Nachweis erbracht, dass die Produktionsstätten nach den Grundsätzen der der GMP-Praxis validiert sind, dass die Geräte, Ausrüstungsgegenstände, Materialien und Reinigungsverfahren implementiert sind, so darf der Hersteller davon ausgehen, dass die Produktionsabläufe tatsächlich die zu erwarteten Ergebnisse liefern.









