
Sicher vor Kontaminationen Reinstverpackungen im Fokus
01.09.2016 -
-
 : Folienrolle „Cleanroom-Master” für den Reinraum. © KWP Kunststoff-Werk-Plur GmbH & Co.KG
: Folienrolle „Cleanroom-Master” für den Reinraum. © KWP Kunststoff-Werk-Plur GmbH & Co.KG -
 orgefüllter Inhalator als Reinraumprodukt © Gerresheimer AG, Düsseldorf
orgefüllter Inhalator als Reinraumprodukt © Gerresheimer AG, Düsseldorf -
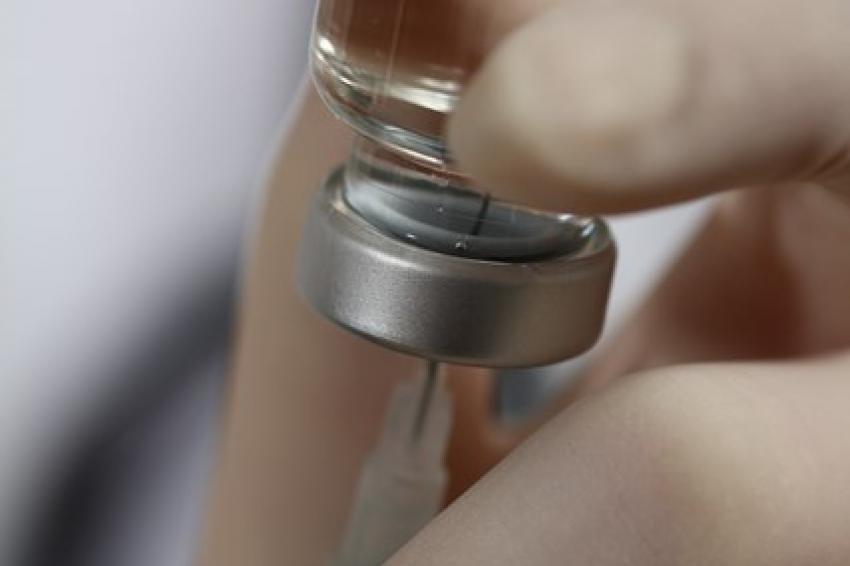 Hightech-Vial aus COP. Solche Vials eignen sich ideal für biopharmazeutische Produkte als Primärpackmittel, da sie leicht und bruchsicher sind. © Gerresheimer AG, Düsseldorf
Hightech-Vial aus COP. Solche Vials eignen sich ideal für biopharmazeutische Produkte als Primärpackmittel, da sie leicht und bruchsicher sind. © Gerresheimer AG, Düsseldorf -
 Fertigspritzen sind Primärpackmittel mit Zusatzfunktion. Hier das ready-to-fill-Set Gx RTF-Spritzen ClearJect aus COP mit Kanüle. © Gerresheimer AG, Düsseldorf
Fertigspritzen sind Primärpackmittel mit Zusatzfunktion. Hier das ready-to-fill-Set Gx RTF-Spritzen ClearJect aus COP mit Kanüle. © Gerresheimer AG, Düsseldorf -
 Die Zytostatika-Schutzverpackung Zyto-Sterilset® dient der Produktsicherheit. © KWP Kunststoff-Werk-Plur GmbH & Co. KG
Die Zytostatika-Schutzverpackung Zyto-Sterilset® dient der Produktsicherheit. © KWP Kunststoff-Werk-Plur GmbH & Co. KG
In der Pharmaindustrie und der Medizintechnik sind Reinraumverpackungssysteme Teil der Prozesskette. Sie sind unerlässlich um Kontaminationsrisiken zu vermeiden. Die Produkte werden in Reinräumen hergestellt und müssen diese anschliessend in steriler Form und in geeigneter Verpackung verlassen. Daher müssen auch die verwendeten Packmittel steril sein und müssen den gleichen Anforderungen genügen wie das im Reinraum hergestellte Produkt.
Reine Verpackungen haben gemäss den regulatorischen Anforderungen eine wichtige Funktion; sie schützen das Produkt vor Kontaminationen, vor Licht, Luft, Feuchtigkeit und mechanischen Beschädigungen und sie können in der täglichen Praxis eine Re-Kontamination der Produkte verhindern.
Verpackungen in der Pharmaindustrie
Verpackungen werden in der ganzen Reinraumprozesskette eingesetzt. Sämtliche Komponenten, vom Rohstoff über das hergestellte Zwischenprodukt bis zum Endprodukt müssen einen Verpackungsprozess durchlaufen.
Wenn die Verpackung dabei unter weniger reinen Bedingungen als das Produkt hergestellt wird, beeinflusst dies unmittelbar das Kontaminationsrisiko das von der Verpackung ausgeht. Alle Hilfsmaterialien welche in Reinräumen verwendet werden, also auch Verpackungen, müssen daher so hergestellt sein, dass eine nachträgliche Kontamination von Produkten in der Weiterverwendung verhindert wird.
Hinsichtlich ihrer Funktion wird zwischen zwischen Primär- und Sekundärverpackungen unterschieden:
Packmittel wie Folien, Gläser, Dosen, Deckel usw., die in direktem Kontakt mit Lebensmitteln oder Arzneimitteln stehen, bezeichnet man als Primärpackmittel. An sie werden besondere Anforderungen gestellt. So muss die Sterilbarriere von Folien z. B. für bis zu 10 Jahren erfüllt sein.
Eine weitere Unterteilung bezüglich der Sterilität ist vor allem in der Pharmaindustrie und Medizintechnik von Interesse. Unter sterilen Primär-Verpackungsformen versteht man Ampullen, Flaschen, Vials, Karpullen und Fertigspritzen. Nicht-sterile Verpackungsformen können Flaschen, Tuben, Dosen, Inhalatoren, Blister und Stickpacks sein.
Sekundärpackmittel dagegen – als äussere Schutzhülle – stehen nicht in direktem Kontakt zum zu verpackenden Gegenstand stehen und haben meist Transportschutz- und Kontrollfunktion.
Verpacken im Reinraum
In Pharmazie und Medizintechnik ist die Herstellung im Reinraum längst Standard. So können die hohen Anforderungen hinsichtlich Sauberkeit und Hygiene erfüllt werden. Das gilt für Wirkstoffe ebenso wie für Bauteile aus Kunststoff, Implantate, Hilfsmittel und Instrumente, aber auch für Verpackungen. Jede Komponente, die in die Reinraumprozesskette eingeschleust wird, muss dabei verpackt sein, damit keine Verunreinigungen aus der Umwelt in den Reinraum gelangen. Am Ende müssen die fertigen Produkte wiederum verpackt werden, um sie partikelfrei aus dem Reinraum zu schleusen.
Die Verpackung muss gemäss den Anforderungen des Kunden unterschiedliche Funktionen erfüllen:
- Schutzfunktion durch Primär- und Sekundärverpackungen
- Transportfunktion zwecks Handling der Produkte im Reinraum
- Informationsfunktion zur Identifikation und zur Qualitätssicherung
Gibt es keine Restschmutzanforderungen, kann durchaus ein einfacher Behälter als Verpackung ausreichen. Bei geringen Beanspruchungen reicht bereits ein Folienbeutel, um Kontaminationen von aussen zu verhindern.
Bei hohen Reinraumanforderungen muss nicht nur die Kontamination von aussen verhindert werden, sondern auch das Erzeugen von Partikeln durch mechanische Belastung der Bauteile oder der Verpackungen z. B. durch Aneinanderreiben. Auch Verschmutzungen durch Abrieb sind bekannt. Das erfordert die richtige Wahl der Containments und des Verpackungsmaterials. Die Packmittel, die eingesetzt werden, sollten möglichst abriebfest sein.
Die Wahl der Verpackung hängt nicht nur von den technischen Spezifikationen, sondern auch von der Bestimmung des Produkts in der Reinraumprozesskette sowie von seiner Sterilisierbarkeit ab.
Um das Kontaminationsrisiko der Produkte bereits bei der Entstehung zu reduzieren, sind kontrollierte Bedingungen und deren dokumentierte Einhaltung für jede Charge erforderlich. Denn Verpackungen müssen nicht nur im Reinraum in jeweils an die Anforderungen angepasst steril produziert werden, auch für Verpackungen muss die volle Rückverfolgbarkeit garantiert sein.
Der Mensch ist die grösste Kontaminationsquelle im Reinraum. Daher müssen die Mitarbeiter grossen Wert auf persönliche Hygiene legen und Gesundheitschecks durchlaufen. Auch die Reinraumbekleidung muss nach Gebrauch gereinigt werden, um das Risiko von Kontaminationen zu vermindern.
Viele Unternehmen verfolgen für ihre Verpackungsanlagen ein Zonenkonzept:
In Zonen der Reinraumklasse A, in denen der Verpackungsprozess abläuft, muss die Umgebung steril sein. In anderen Bereichen der Verpackungsanlage muss sie nicht steril sein. Um Verwechslungen zu vermeiden, sind verschiedene Aufträge in der Verpackungsanlage strikt voneinander zu trennen.
Dabei muss eine Kontamination durch die eingesetzten Verpackungsmaschinen und Geräte ausgeschlossen werden können. Nach dem Verpackungsprozess wird deshalb oft noch ein zusätzlicher Sterilisationsschritt, z. B. mittels UV-oder Röntgenlicht, eingefügt.
Zum Nachweis, dass die Toleranzwerte für die Luft und die Oberflächen eingehalten wurde, werden die dafür erforderlichen Parameter permanent erfasst.
Im Verpackungsprozess werden daher oft Automationslösungen favorisiert, die die Kontaminationsquelle Mensch ausschliessen und die Prozesssicherheit zu optimieren. Auch das richtige Design ist in der Reinraumtechnik enorm wichtig. Auf diesem Gebiet hat man in letzter Zeit grosse Fortschritte gemacht.
Kontaminationen
Die Problematik der Kontamination durch Prozess-Hilfsstoffe und Abriebmaterial wird heute immer noch unterschätzt. Dabei ist z. B. die Gefahr der Übertragung von Silikon-Öl-Rückständen hoch. Und bereits eine Monoschicht von Silikonöl-Rückständen auf einem Implantat kann bewirken, dass ein Implantat vom Körper abgestossen wird. Auch Staubpartikel, die von Verpackungen herrühren, können Kontaminationen bewirken.
Der Nachweis der Reinraumtauglichkeit ist durch den Hersteller der Verpackungsanlage anhand einer professionellen Qualifizierung zu erbringen. Im laufenden Betrieb sind allerdings regelmässige Kontrollen, Requalifizierungen, erforderlich um einen ordnungsgemässen Betrieb zu garantieren.
Welches sind die Standards für den Nicht-Steril-Betrieb? Viele Entscheidungen sind heute nicht durch Normen vorgegeben, sondern sie liegen in der Verantwortung des Produzenten. Wenn dieser sich für einen Nicht-Steril-Betrieb entscheidet, muss er ihn auch vor den Auditoren verteidigen können. Insektenbefall, Staubbildung und eine Kreuzkontamination der Produkte müssen ausgeschlossen werden können. Die Lüftung muss den Normen entsprechen. Der Hersteller muss den Nachweis erbringen, dass das Produkt sicher ist.
Indes geht der Trend hin zu sterilen Fertigarzneimitteln, wie etwa Fertigspritzen, insbesondere bei hochpreisigen Produkten aus dem biotechnologischen Bereich, die vorverpackt zum Patienten gelangen. Innerhalb der Fertigspritzen geht der Trend klar hin zu gebrauchsfertigen Spritzen (Ready-to-fill – RTF).
Normative Anforderungen an Reinraumverpackungen
Gesetze und Normen dienen der Vermeidung von Risiken. In der gesamten Prozesskette, vom Kundenauftrag über die Produktion, die Verpackung bis hin zum Versand, muss der Produzent in der Lage sein, seine GMP-gerechte Produktion nachzuweisen. Der Aufbau eines dafür erforderlichen Qualitätsmanagementsystems, das sämtliche Prozesse unterstützt, ist nicht einfach und erfordert einen permanenten Lernprozess aller Mitarbeiter.
Die Verpackung von Medizinprodukten ist dafür vorgesehen, die Sterilität der Endverpackung bis zum Anwendungszeitpunkt zu erhalten; in diesem Umfeld sind dabei folgende Normen gültig: Für Anforderungen an Materialien, Sterilbarrieresysteme und Verpackungssysteme die DIN EN ISO 11607-1, für Validierungsanforderungen an Prozesse der Formgebung, Siegelung und der Komposition der Verpackungen die DIN EN ISO 11607-2. Für Biomaterialien gilt der ASTM F2475 – 11 Standard Guide for Biocompatibility Evaluation of Medical Device Packaging Materials.
Die DIN EN 868-2 regelt die Prozesse für Sterilisierverpackung -– Anforderungen und Prüfverfahren für Verpackungen enthält Prüfverfahren und Materialangaben für sterilisierte Medizinprodukte. Für Arzneimittelverpackungen gilt die DIN 15378 (Primärverpackungen für Arzneimittel).
Fälschungssicherheit
Pharmafälschungen werden zu einer immer grösseren Bedrohung für den Pharmamarkt und für die Anwender. Die Weltgesundheitsorganisation schätzt, dass rund 25 % der in Entwicklungsländern eingenommenen Medikamente gefälscht sind. Die gefälschten Medikamente kommen selbst gegen lebensbedrohliche Krankheiten wie Tuberkulose und AIDS auf den Markt und schädigen die Verbraucher – bis hin zu Todesfällen.
Solche Fälschungen müssen über Verpackungsspezifikationen bei der Etikettierung mit speziellen Siegeln und Codes verhindert werden. Seit 2015 (ab 2017 absolut verbindlich) darf gemäss Fälschungsrichtlinien der Europäischen Union allein wegen dieser Produktpiraterie, aber auch zur Produktsicherheit, keine Charge mehr ohne Seriennummer ausgeliefert werden. Das Aufbringen von Seriennummern, nicht mehr nur auf komplette Chargen, sondern auf alle einzelnen verkaufsfähigen Packungen sowie die folgenden Verpackungsstufen wie Karton oder Palette, dient gleichzeitig der Sicherheit des Patienten wie der Abwehr der Produktpiraterie. Jede Charge erhält eine Etikette mit Herstellerangabe, Materialbezeichnung, Typ und Artikelnummer, Datum und Lotnummer. Auf diese Weise kann das Produkt jederzeit eindeutig identifiziert und rückverfolgt werden.
Um im Pharmaprozess die erforderliche Flexibilität und Transparenz zu garantieren, wurden neue technische Lösungen für Serialisierungssysteme zur Integration des Etikettendrucks in die ERP-Systeme und in die Auftragsverarbeitung entwickelt. Für den Produzenten steht dabei eine rasche und fehlerfreie Kennzeichnung unterschiedlicher Produkte mit 2D-Matrix-Codes im Vordergrund. Moderne Etikettiersysteme sind mit einer Kontrollfunktion, d. h. einer gleichzeitigen Verifizierung und Prüfung der Lesbarkeit sowie einer dynamischen Anpassbarkeit an Veränderungen ausgestattet.
Anforderungen an die Packmittel
Im Medizintechnikbereich müssen alle Materialien u. a. Bio-kompatibel sein, schon allein wegen möglicher Allergie- und Entzündungsgefahren durch Verunreinigungen, die von Packmitteln ausgehen können. Verpackungsmaterialien müssen eine sehr hohe chemische und partikuläre Reinheit aufweisen und eine hohe Stabilität gegenüber Umwelteinflüssen aufweisen. Ein Standardguide für die biologische Evaluation schreibt vor, dass über eine ID die Biokompatibilität gewährleistet sein muss. Denn pharmazeutische Packmittel sind mehr als nur ein Behältnis für die Formulierung. Gerade bei Biopharmazeutika haben sie einen nicht zu unterschätzenden Einfluss auf Qualität, Wirksamkeit und Unbedenklichkeit wie auch auf die unbedenkliche Anwendbarkeit des Arzneimittels.
Das Produkt und seine Verpackung müssen optimal aufeinander abgestimmt sein, denn die Primärverpackung und das Produkt stehen in direktem Kontakt und Verpackungsmaterialien – wie etwa Weichmacher und Radikalfänger – können für den Anwender zum Problem werden. Wenn die Verpackung das Produkt beeinflusst, dann sollte man sie daher austauschen.
Falls in einem Reinraum gearbeitet wird, können für die Innen- oder Primärverpackung auch Folien verwendet werden, die selbst im Reinraum produziert und konfektioniert wurden und eine definierte Partikellast aufweisen. Auch eine antistatische Ausrüstung von Folien kann eine Kontamination mit Staub reduzieren.
Während der Lagerung ist eine Migration von Sauerstoff durch die Verpackung hindurch möglich. Durch den Einsatz Sauerstoffdichter Verpackungen kann jedoch eine Oxidation der Produkte verhindert werden, indem kein weiterer Sauerstoff aus der Atmosphäre in das Produkt eindringen kann.
Wenn lichtempfindliche Materialien verpackt werden müssen, kann eine geeignete Verpackung (bspw. farbiges oder UV-undurchlässiges Glas) das Produkt vor Lichteinwirkung schützen.
Lieferkette
Was geschieht mit Versandverpackungen beim Transport mit Lkw, Flugzeug oder Schiff? Gerade hier ist die Unsicherheit besonders gross. Daher ist eine permanente Überwachung der Verpackungen in der Logistikbranche – auch wegen der Garantiefunktion – ein grosses Thema.
Beim Um- und Abladen kommt es bei unzureichender Verpackung immer wieder zu Beschädigungen und damit zu Rückverschmutzungen. Je nach Transportart sollten daher mechanisch belastbare Verpackungen gewählt werden. Gegebenenfalls muss der Hersteller über stabilere Sekundärverpackungen zum Schutz gegen aussen nachdenken.
Verfügt der Produzent der Verpackungsmaterialien über ein qualifiziertes Qualitätsmanagement und eine ISO-Zertifizierung? Ein Lieferantenaudit schützt vor Reklamationen: Primärpackmittel benötigen immer ein Audit, das alle 3 Jahre zu wiederholen ist (gemäss ISO 15378)
Neuheiten
Wenn im Reinraum die Produkte mehrstufige Prozesse durchlaufen, sind ineinander steckbare Sacksysteme interessant. Der Anwender öffnet diese und befüllt den inneren Beutel mit dem Produkt. Dann werden die verschiedenen Beutel um das Produkt herum einzeln verschweisst und können später in den einzelnen Reinraumzonen sukzessive wieder abgenommen werden. Dieses bag-in-bag-System ist ideal für das GMP-konforme Einschleusen in den Reinraum. Solche „Zwiebelschalen“-Systeme bieten z. B. die Firma Strubl, Wendelstein, oder die Firma KWP Plur, Gelnhausen, an. Auf diese Weise lässt sich das komplizierte einzelne Abpacken in mehreren Schichten ersparen.
Neuartige Verpackungen, die neben einer reinen Umhüllungsfunktion und dem Kontaminationsschutz weitere Aufgaben haben, finden vermehrt Anwendung in der Medizintechnik. Ein typisches Beispiel sind vorgefüllte Spritzen (Fertigspritzen), die neben der Behältnisfunktion auch zur Applikation des Arzneimittels verwendet werden. Gx RTF ClearJect Spritzen aus COP anstelle von Glas, die demnächst auf den Markt kommen, sind besonders bruchfest und daher gut für die Verpackung aggressiver oder toxischer Wirkstoffe geeignet.
Ein wichtiger Ansatz in der Verpackungsbranche beinhaltet, sämtliche Kontaminationsrisiken schon während der Produktion auszuschliessen, so dass die nachträgliche Sterilisation entfallen kann.
Zytostatika sind Krebs- und Autoimmunmedikamente, die das Zellwachstum hemmen. Da sie hochwirksam sind, sollten sie nicht in die Umgebung gelangen. Sterilisierte Zytostatika werden deshalb in einer Zytostatika-Schutzverpackung für Transport, Lagerung und für den Transport angeboten. Das System ermöglicht ein sicheres Verschliessen und Öffnen und ist GMP-kompatibel.









