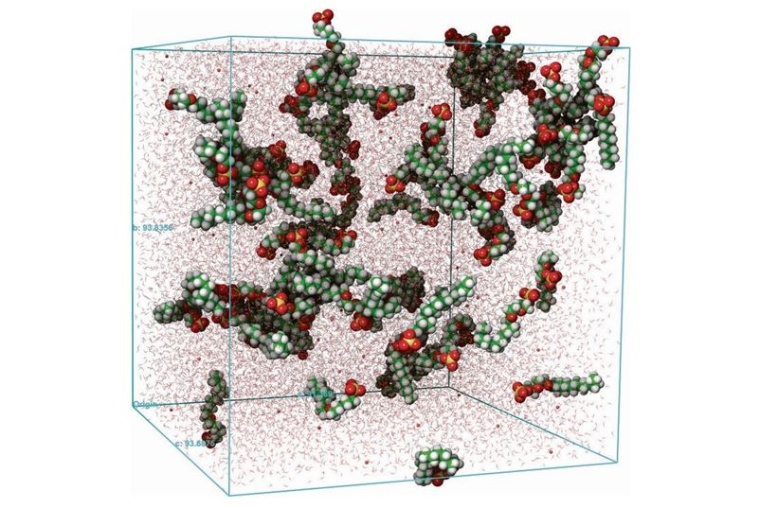
Digitales Moleküldesign
Computergesteuertes Moleküldesign mit seinen Möglichkeiten, riesige Mengen simulierter Daten zu erzeugen, verschafft Zugang zu neuen Bereichen chemischer Forschung für neue Materialien.
Ein gewaltiges Erdbeben zerstörte 17.000 Häuser, die umfallenden Kerzen entfachten Brände in der ganzen Stadt, ein Tsunami tötete die letzten Überlebenden am Strand und die anschließenden Plünderungen vervollständigten das apokalyptische Szenario in Lissabon am 1. November 1755.
Die Bestürzung in ganz Europa nach dieser Katastrophe war enorm; Voltaire veröffentlichte sein „Poème sur le désastre de Lisbonne“, in dem er mit dem Optimismus einer an sich weisen Welt bricht. Dieser Wandel in der Sichtweise beschleunigte die Entwicklung modernen Denkens — die europäische Aufklärung –, die zu einem elementaren Prinzip moderner Wissenschaft wurde.
Der Prozess der Verwissenschaftlichung der Gesellschaft ermöglicht die Prävention von Katastrophen durch Antizipation von Lösungen basierend auf datengestützter Vorwegnahme des Kommenden. Traditionell stützt sich dieser Prozess ausschließlich oder in erheblichem Maße auf experimentelle, empirische Daten. Mit der exponentiellen Beschleunigung und Skalierbarkeit von Supercomputern sind wir nun jedoch in der Lage, simulierte in-silico-Daten mit einer Genauigkeit zu erzeugen, die experimentellen Methoden entspricht, jedoch in einem noch nie dagewesenen Umfang. Wir stehen vor einer nicht nachhaltigen Zukunft, aber wir haben das Potenzial moderner Wissenschaft gepaart mit der Leistungsfähigkeit von Computersimulationen, um den Kurs zu ändern.
Neue Materialien für den „Green Deal“
Mit dem Ziel, eine nachhaltigere und kreislauforientierte Wirtschaft aufzubauen, hat die Europäische Union den „Green Deal“ unterzeichnet. Dieser ist zwar in seiner Wirkung potenziell revolutionär, stellt aber eine Umwälzung für die gesamte chemische Prozesskette und die damit verbundenen Branchen dar. Chemikalien, die jahrzehntelang verwendet wurden, können nicht mehr eingesetzt werden, und die Hersteller stehen vor der Herausforderung, nachhaltige Ersatzstoffe zu finden. Wir brauchen neue Materialien, und wir brauchen sie schnell. Hier verspricht wissenschaftlicher Fortschritt schnellere Antworten als in der Vergangenheit.
Computergesteuertes Moleküldesign mit seinen Möglichkeiten, riesige Mengen simulierter Daten zu erzeugen, verschafft Zugang zu neuen Bereichen chemischer Forschung für neue Materialien. Es verspricht Schnelligkeit und Genauigkeit und ermöglicht uns, große molekulare Räume zu durchforsten, um gezielt die vielversprechendsten Moleküle auszuwählen und experimentell zu validieren.
Die potenziellen Methoden für computergesteuertes Moleküldesign sind vielfältig, und die Algorithmen werden seit Jahrzehnten von der Wissenschaft kontinuierlich weiterentwickelt. Die Ansätze können in zwei Kategorien unterteilt werden:
- Wissensbasiertes maschinelles Lernen, das heute oft als künstliche Intelligenz (KI) bezeichnet wird. Es hat in letzter Zeit viel Aufmerksamkeit erlangt, weil es Menschen beim Schach und beim Go schlagen, Gesichter auf Fotos fast so gut erkennen und Autos fast so gut fahren kann wie Menschen — da stellt sich natürlich die Frage: Wie gut ist es im Entwickeln neuer Moleküle?
- Stringente, auf grundlegenden physikalischen Prinzipien basierende Modelle, welche die den molekularen Eigenschaften zugrunde liegende Physik genau beschreiben. Dies erfordert große Fortschritte in der Beschreibung molekularer Wechselwirkungen als auch bei der Rechenleistung.
Wenn wir ein Entwicklungsverfahren anstreben, das sowohl einen großen chemischen Raum abdeckt als auch Präzision auf experimenteller Ebene bietet, dann ist hierzu das Zusammenspiel von maschinellem Lernen und Physik der Schlüssel. Hinsichtlich der Rechenzeit ist es schneller, Milliarden von Molekülen zu konstruieren und sie dann entweder mit wissensbasierten Algorithmen oder einfachen physikalischen Methoden herauszufiltern. Die nach diesem Filterprozess übrig gebliebenen Moleküle werden dann mit den zuverlässigsten physikalisch basierten Methoden berechnet, um Ergebnisse mit experimenteller Genauigkeit zu erhalten. Die besten Verbindungen können dann wieder als Grundlage für die Generierung von Molekülderivaten dienen, und dieser Zyklus kann wiederholt werden, bis eine Reihe von Verbindungen mit den gewünschten molekularen Eigenschaften berechnet ist.
Nicht nur das Zusammenspiel zwischen den verschiedenen Berechnungsmethoden ist entscheidend, sondern auch das zwischen experimentellen und in-silico-Daten. Die Fähigkeit, beide Datentypen in ihrer Gesamtheit zu interpretieren und in-silico-Daten auf der Grundlage bekannter experimenteller Daten zu generieren, ist zentral. Außerdem führt die Interpretation experimenteller Daten mit Simulationsmethoden zu einer neuen Art von Unschärferelation, bei der die Interpretation von experimentellen und in-silico-Daten nicht mehr voneinander getrennt werden kann. Laborautomatisierung und Kontextualisierung großer Datenmengen, sowohl experimentell als auch in silico, gehen Hand in Hand. Die Grundsätze menschlicher Entscheidungsfindung in modernen Wissenschaften bleiben jedoch die gleichen, nur die Art der zugrunde liegenden Daten hat sich geändert.
Neue Bereiche grüner Chemie
Einige Beispiele für das computergestützte molekulare Design von Materialien zeigen, dass wir durch diesen Ansatz in neue Bereiche grüner Chemie vorstoßen können:
Auf der International Elastomer Conference in Pittsburgh präsentierte Atif Afzal von Schrödinger eine Zusammenarbeit mit Evonik zum Thema „Using Molecular Simulation to Assess the Impact of Additives and Macrocyclic Structures on Vestenamer“. Evonik nimmt eine Vorreiterrolle in der Kreislaufwirtschaft ein. Eines der Produkte des Unternehmens ist Vestenamer, das als Prozessadditiv für das Gummirecycling verwendet werden kann. In Europa fallen jährlich 3,6 Mio. t Altreifen an. Um diese riesige Menge zu recyceln, werden Produkte wie Vestenamer benötigt.
Evonik und Schrödinger untersuchten gemeinsam das molekulare Verhalten von Vestenamer bei der Anwendung im Gummirecycling. Simulationstechnologien können helfen, den Prozess auf molekularer Ebene zu verstehen, so dass die optimalen Verarbeitungsbedingungen und weitere Anwendungsbereiche gefunden werden können. In dieser speziellen Arbeit wurden die Wechselwirkungen von Vestenamer mit verschiedenen Kautschukpolymeren und typischen Vernetzungsadditiven analysiert, so dass strukturelle und thermophysikalische Eigenschaften vorhergesagt werden konnten. Die Arbeit lieferte auch Erkenntnisse darüber, wie Vestenamer dazu beiträgt, verschiedene Kautschukpolymere miteinander zu verbinden, was wiederum zu einer besseren Wiederverwertbarkeit der Mischung führt.
Beim User Group Meeting von Schrödinger im September 2021 präsentierten Mariam Hussain und Martin Settle von Reckitt in ihrem Vortrag „Accelerating Formulation and Packaging Development Using Molecular Dynamics“, wie digitale Methoden helfen können, neue Konsumgüter schneller zu entwickeln. Gerade in diesem Umfeld haben der Green Deal und die Nachfrage nach nachhaltigeren Produkten einen starken Einfluss auf den Innovationsprozess. Welches neue biobasierte Tensid ist das beste für eine bestimmte Anwendung? Unternehmen wie Reckitt müssen einen Weg finden, um neue Chemikalien und Materialien zu bewerten und zu prüfen, ob sie für ein bestimmtes Produkt geeignet sind.
Hussain und Settle zeigten, wie Simulationen bei Reckitt eingesetzt werden, um neue Ideen für Produktformulierungen und Verpackungen digital zu bewerten. Neue Inhaltsstoffe wie Tenside oder Polymere werden mit Hilfe von Simulationsansätzen auf eine bestimmte Eigenschaft und ein bestimmtes Verhalten hin überprüft, und die besten Kandidaten werden anschließend getestet. Das Herausfiltern der besten Kandidaten, die geprüft werden sollen, anstatt alle Kandidaten zu testen, ist entscheidend für die Beschleunigung des Designprozesses, da ein experimenteller Test für ein neues Produkt oder eine neue Verpackung Monate in Anspruch nehmen kann. Ein besonderes Beispiel war die Auswahl neuer Tenside für Waschmittel und deren Wechselwirkung mit Beschichtungen auf Textilien. Mit Hilfe von Simulationsansätzen konnten die Reckitt-Wissenschaftler neue Tenside, die von ihren Lieferanten angeboten wurden, bewerten und die besten für eine bestimmte Anwendung heraussuchen. Im Durchschnitt beschleunigen solche digitalen Ansätze den Innovationsprozess um das Zehnfache im Vergleich zu rein experimentellen Methoden.
In seiner Funktion als Direktor für Nachhaltigkeit betonte Settle auch, dass Molekülsimulationen bei Reckitt für die Beschleunigung von Innovationen von entscheidender Bedeutung sind, insbesondere für die Einführung neuer, nachhaltigerer chemischer Verfahren für Produkte und Verpackungen. Bis 2030 will Reckitt 50 % des Nettoumsatzes mit nachhaltigeren Produkten erzielen.
Die Vorteile molekularer Simulationen sind nicht auf große chemische Unternehmen beschränkt. Simulationen sind auch für kleinere Firmen und agile Start-ups hilfreich, um Innovationen zu beschleunigen. Hier sind prädiktive Ansätze von entscheidender Bedeutung, um neue Materialien oder neue Anwendungsbereiche schnell zu erforschen. Ein aktuelles Beispiel wurde von Cambium Biomaterials, einem in Kalifornien ansässigen Start-up für Biopolymere, vorgestellt. Andrew Guenthner, Chief Technology Officer, erläuterte, wie die Technologie von Schrödinger eingesetzt wird, um neue biobasierte Polymere für Flammschutzmittel in der Luft- und Raumfahrt zu entwickeln, die eine bessere Leistung aufweisen als die traditionellen rohölbasierten Produkte. Biobasierte Ausgangsstoffe liefern neue Monomereinheiten, die im Vergleich zu den traditionellen rohölbasierten Bisphenolverbindungen einen wesentlich größeren Gestaltungsspielraum für Epoxidharze bieten. Das Unternehmen nutzte Simulationen beim Screening von Zieleigenschaften wie der Glasübergangstemperatur oder dem mechanischen Verhalten in Abhängigkeit von der molekularen Zusammensetzung und den Verarbeitungsbedingungen. Auch in diesem Fall führte der digitale Ansatz zum Screening einer Bibliothek mit verschiedenen Kandidaten zu einem beschleunigten Innovationsprozess.
Unsere Gesellschaft fordert zu Recht, dass industrielle Produktion nachhaltiger und kreislauffähiger werden muss, um eine apokalyptische Zukunft zu verhindern. Moderne Wissenschaft und insbesondere digitales Moleküldesign können eine entscheidende Rolle in diesem industriellen Transformationsprozess hin zu einer Zukunft spielen, in der wir alle gerne leben.
Autor:
Jörg Weiser, Geschäftsführer, Schrödinger GmbH, München
_______________________________________________
Digital Chemistry & Materials Innovation
In einer Vortragsreihe von Schrödinger in Kooperation mit CHEManager können Sie mehr zur digitalen Transformation in der Chemie und Materialwissenschaft erfahren:
https://bit.ly/MolecularModelling.