
Komplette Transparenz
EU-Initiative gegen Medikamentenfälschungen erreicht nächsten Meilenstein
-

-
 Abb. 1: Point-of-Dispense-Verifikationsmodell. Die gestrichelten Pfeile deuten an, dass die Großhändler in der Lage, aber nicht verpflichtet sind, die Arzneimittelpackungen zu verifizieren.(© EFPIA)
Abb. 1: Point-of-Dispense-Verifikationsmodell. Die gestrichelten Pfeile deuten an, dass die Großhändler in der Lage, aber nicht verpflichtet sind, die Arzneimittelpackungen zu verifizieren.(© EFPIA) -
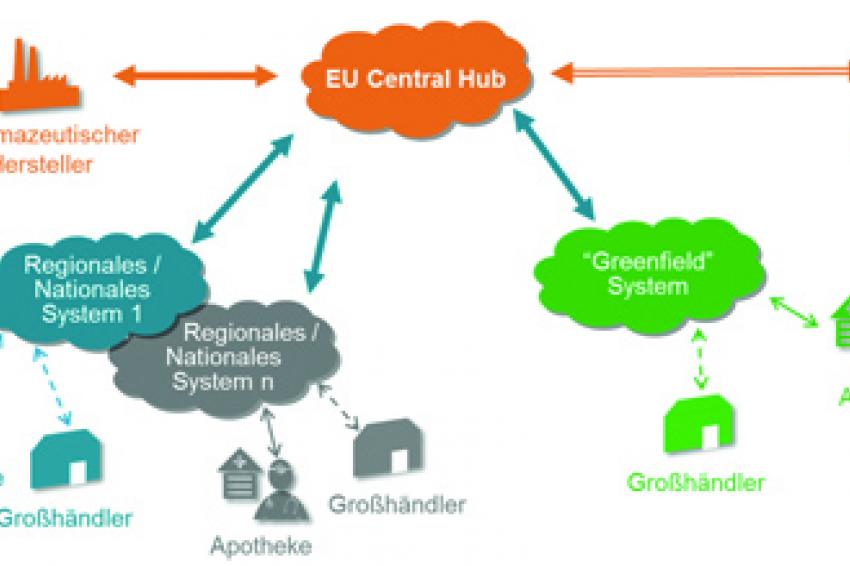 Abb. 2: Systemlandschaft für ein europäisches Arzneimittelverifikationssystem(© EFPIA)
Abb. 2: Systemlandschaft für ein europäisches Arzneimittelverifikationssystem(© EFPIA) -
 Dr. Stefan Artlich, Bayer Technology Services
Dr. Stefan Artlich, Bayer Technology Services
Im Kampf gegen den besorgniserregenden Anstieg der Zahl gefälschter Arzneimittel haben Europäisches Parlament und EU-Ministerrat im vergangenen Jahr die Richtlinie 2011/62/EU erlassen. Kerninhalte aus Sicht der pharmazeutischen Industrie sind die Festlegung von Sicherheitsmerkmalen zur Überprüfung der Echtheit von Arzneimitteln und die Einführung eines Systems zur Verifizierung einzelner Medikamentenpackungen.
Grundsätzlich werden alle verschreibungspflichtigen Arzneimittel diese Sicherheitsmerkmale tragen müssen, es sei denn, sie finden den Weg auf die ‚whitelist‘ der Ausnahmen. Bei den nicht verschreibungspflichtigen Produkten ist es genau umgekehrt: Sie sind von der Regelung bzgl. Sicherheitsmerkmalen ausgenommen, es sein denn, sie finden sich auf der ‚blacklist‘ wieder, z. B. aufgrund einer entsprechenden Bewertung des Fälschungsrisikos. Die Herstellung der eingesetzten Wirkstoffe muss den Grundsätzen der guten Herstellungspraxis (GMP) genügen, auch wenn sie außerhalb der EU produziert werden. Dies soll u. a. durch Audits bei den Wirkstoffherstellern, aber auch durch schriftliche Bestätigung der Arzneimittelhersteller, die ihrerseits die Einhaltung bei ihren Wirkstoff-Lieferanten überprüfen müssen, sichergestellt werden.
Konzeptpapier veröffentlicht
Während die Richtlinie das Thema GMP für Wirkstoffe umfangreich beschreibt, sollen Details zu den übrigen Themen durch die EU-Kommission im Rahmen von „delegierten Rechtsakten" festgelegt werden. Zur Vorbereitung hierauf hat die Kommission im November 2011 ein Konzeptpapier veröffentlicht und alle Stakeholder zur Kommentierung eingeladen. In dem Dokument werden zu einzelnen Themengebieten verschiedene Szenarien skizziert, zum Teil bereits mit Vor- und Nachteilen bewertet und dann konkrete Fragen gestellt.
Die Option, die Art der Produktkodierung jedem Hersteller selbst zu überlassen, ist hierbei vermutlich eher der Vollständigkeit halber erwähnt, würde sie doch zu einer schier unüberschaubaren Kodierungslandschaft in Europa führen. Eine diesbezügliche Festlegung ist daher dringend geboten und muss zumindest eine Produktnummer und eine randomisierte Seriennummer enthalten. Batchnummer und Verfalldatum sind zwei denkbare weitere Bestandteile einer Produktkodierung, für deren Nutzung sich bereits der Europäische Verband der forschenden Arzneimittelhersteller (EFPIA) gemeinsam mit den europäischen Verbänden der Apotheker (PGEU), Großhändler (GIRP) und Parallelimporteure (EAEPC) ausgesprochen hat. Und auch bei der Frage, ob die Produktkodierung in Form eines linearen Barcodes, eines 2D-Barcodes oder eines RFID-Chips vorgenommen werden soll, votieren EFPIA und die übrigen Verbände gemeinsam, und zwar für den zweidimensionalen DataMatrix-Code.
Verifikation von Arzneimitteln
In Erwartung des zurzeit laufenden europäischen Gesetzgebungsverfahrens hat die EFPIA das Point-of-Dispense-Verifikationsmodell entwickelt, das auf der Kodierung jeder einzelnen Arzneimittelpackung mit Produktnummer, zufallsgenerierter Seriennummer, Batchnummer sowie Verfalldatum basiert. Diese Daten werden vom Hersteller per DataMatrix-Code auf jede produzierte Packung gedruckt und gleichzeitig in eine zentrale Datenbank übertragen. Bei der Abgabe der Arznei in der Apotheke scannt der Apotheker den Code, und sein Kassensystem startet im Hintergrund eine Anfrage an die zentrale Datenbank. Ist eine Packung mit den eingescannten Daten dort bekannt, so wird sie an den Patienten ausgehändigt und gleichzeitig als „verkauft" markiert. Schlägt die Datenbankabfrage dagegen fehl, so weiß der Apotheker, dass er eine Packung in den Händen hält, die nicht an den Patienten abgegeben werden darf.
Dieses Verifikationsmodell wurde 2009/2010 in einem Pilotprojekt der EFPIA in ausgewählten schwedischen Apotheken erfolgreich im operativen Tagesgeschäft getestet und passt zu dem, was in dem Konzeptpapier der EU skizziert wird: Das „Auschecken" der Packung im System bei Abgabe an den Patienten muss verpflichtend sein.
Eine aus Herstellersicht bedeutsame Festlegung ist bereits in der Richtlinie 2011/62 enthalten: Die Kosten für das zentrale Datenbanksystem werden die Hersteller tragen müssen. Doch wie könnte die Architektur für ein solches System für eine EU mit gegenwärtig 27 Mitgliedsstaaten überhaupt aussehen, und wie die dazugehörige Organisation? Zum zweiten Punkt zieht das vorliegende Konzeptpapier drei Varianten in Betracht: Bei einer Stakeholder-Organisation würden die delegierten Rechtsakte die Ziele für ein Verifikationssystem sowie die Pflichten der einzelnen Stakeholder festlegen und alles weitere wie den Aufbau und Betrieb des Systems sowie die Festlegung der Regeln zur Nutzung gemeinschaftlich in die Hände von pharmazeutischen Herstellern, Großhändlern und Apothekern legen. Als weitere Varianten für die genannten organisatorischen Aufgaben zieht das Konzeptpapier die Governance durch die EU-Kommission oder Europäische Arzneimittelagentur (EMA) sowie durch nationale Behörden („national bodies") in Betracht.
Systemarchitektur und Organisationsfragen
Zur Frage der Systemlandschaft hat die EFPIA gemeinsam mit den übrigen Stakeholder-Verbänden einen Vorschlag entwickelt: Das Gesamtsystem besteht aus einem EU Central Hub und angeschlossenen nationalen Systemen. Der EU Central Hub bietet Herstellern und Parallelimporteuren die Möglichkeit, ihre Packungsdaten zentral über einen einzigen Zugang in die verschiedenen nationalen Systeme der EU-Mitgliedsstaaten zu übermitteln. Die eigentliche Produktverifizierung und das Auschecken der Packung beim Verkauf erfolgt dann pro Land gegen den Datenbestand im jeweiligen nationalen System.
Für die Entwicklung der nationalen Systeme sind verschiedene Varianten denkbar: Stakeholder-Organisationen der Länder können sich entscheiden, ihr eigenes System zu entwickeln, das z. B. perfekt in eine vorhandene IT-technische Apotheken-Infrastruktur eingebettet ist, müssen dafür jedoch das notwendige Know-how aufbauen und die Kosten für Systemdesign und -entwicklung allein aufbringen. Es können sich aber auch Länder zusammenschließen, um ein gemeinsames System zu entwickeln und zu betreiben. Und schließlich ist mit den „Greenfield Systems" eine Variante angedacht, bei der Systementwicklung und -betrieb von einer europäischen Stakeholder-Organisation im Auftrag einzelner nationaler Organisationen übernommen werden.
Damit wird klar: Von den im Konzeptpapier aufgeführten drei Organisationsvarianten präferieren Hersteller, Großhändler und Apotheker die Stakeholder-Variante. Dazu Dr. Martin Friedrich, Head of Product Tracking and Authentication bei Bayer Technology Services und gleichzeitig Program Manager bei der EFPIA: „Für den Erfolg unserer Arbeit ist es wichtig, dass alle Partner gemeinsam an einem Tisch sitzen. Die etwaige Sorge, dass die Pharmaindustrie aufgrund der Verpflichtung zur Übernahme der Kosten originär eigene Interessen durchsetzen könnte, ist daher vollkommen unbegründet."
Der richtige Zeitpunkt ist jetzt ...
Wie geht es nun weiter? Wenn die EU-Kommission Ende April die Stellungnahmen zu dem im November veröffentlichten Konzeptpapier erhalten hat, werden die Kommentare und Antworten auf den Internetseiten der EU veröffentlicht. Sie sind Input für die weitere Arbeit der Kommission, die über verschiedene Stufen in der Veröffentlichung der delegierten Rechtsakte im Jahr 2014 mündet. Anschließend haben die EU-Mitgliedsstaaten Zeit, diese Rechtsakte innerhalb von drei Jahren in nationales Recht umzuwandeln. Ein scheinbar langer Zeitraum, doch die Erfahrungen mit den Serialisierungsanforderungen des türkischen Gesundheitsministeriums sowie der Einführung der nicht-serialisierten DataMatrix-Kodierung in Frankreich zeigen, dass auf die pharmazeutischen Hersteller eine Menge Arbeit zukommt. Dazu nochmals Dr. Friedrich: „Jede Firma benötigt zunächst ein Gesamtkonzept, das die vorhandene IT-Infrastruktur sowie alle die EU beliefernden Verpackungsstandorte mit ihrer erfahrungsgemäß gewachsenen Anlagenvielfalt berücksichtigt. Für die Umsetzung sind dann mittelfristige Investitionsplanung anzupassen und geeignete Lieferanten zu beauftragen. Wer da zu spät beginnt, wird am Ende nicht pünktlich fertig sein, denn schon jetzt besteht ein Wettbewerb um die knappen Ressourcen im Markt."










