
Strategie & Management
Medizintechnik: Von der Idee zum Produkt
Medizintechnikprodukte gesetzeskonform planen, entwickeln und erfolgreich zulassen
-
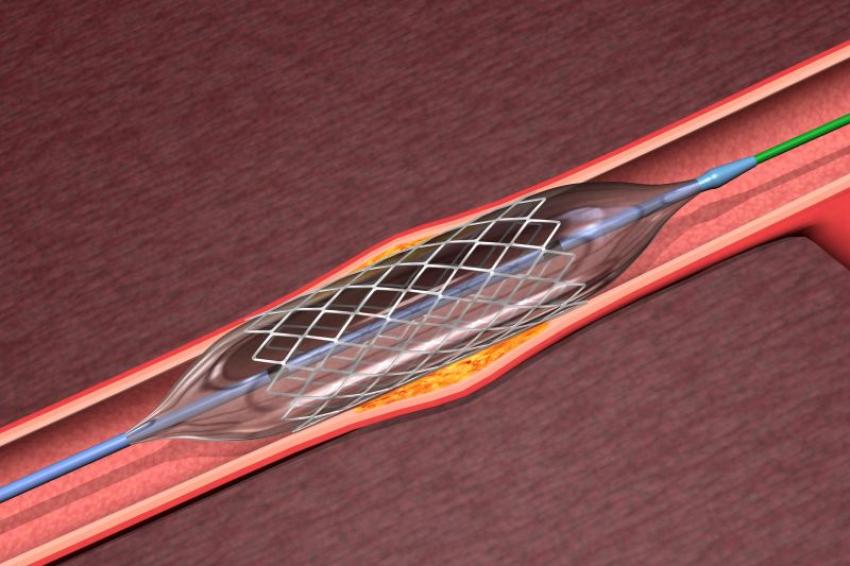 Mit Hilfe von biochemischen Veredelungsmethoden wurden Implantate in den letzten 15 Jahren immer mehr zu einer Art Multitalent in den Händen der Ärzte und im Körper des Patienten. (© BioMedical/Shutterstock)
Mit Hilfe von biochemischen Veredelungsmethoden wurden Implantate in den letzten 15 Jahren immer mehr zu einer Art Multitalent in den Händen der Ärzte und im Körper des Patienten. (© BioMedical/Shutterstock) -
 Dietmar Schaffarczyk, CEO, Stimos GmbH, Konstanz
Dietmar Schaffarczyk, CEO, Stimos GmbH, Konstanz
Vorbei sind die Zeiten, als ein Implantat ein „einfaches“ Ersatzteil war, das mit geprüften mechanischen Eigenschaften Funktionen eines degenerierten Gelenks unterstützen oder ersetzen sollte. Medizintechnikprodukte, Implantate wie auch Instrumente und Apparate, werden immer komplexer und zeichnen sich neben ihrer Primärfunktion durch sekundäre – jedoch nicht minder sicherheits- und leistungsrelevante – Eigenschaften aus. Die klassische Medizintechnik schaut längst über den Tellerrand hinaus und kombiniert ihre Produkte mit Eigenschaften, die oftmals der Forschung und Entwicklung in der Biochemie oder Pharmazie entspringen. Medical Devices sollen heutzutage „smart“ sein, speziell auf den Patienten zugeschnitten oder mit Eigenschaften versehen werden, die ein Implantat zum Alleskönner machen.
Analysiert man den Markt der Implantate, so erkennt man schnell: Die Biochemie hat auf den meisten Implantatoberflächen bereits ihre Spuren hinterlassen. Seien es künstliche Hüften, Zahnimplantate, Wirbelsäulenimplantate oder Stents: Mit Hilfe von biochemischen Veredelungsmethoden wurden Implantate in den letzten 15 Jahren immer mehr zu einer Art Multitalent in den Händen der Ärzte und im Körper des Patienten: Dank biochemischer Beschichtungen wachsen Implantate schneller ein, werden von der Immunabwehr des Patienten nicht als Fremdkörper wahrgenommen, setzen Wirkstoffe genau dosiert und genau an der richtigen Stelle im Körper frei und wirken antibakteriell oder entzündungshemmend.
Komplexe Wirkweise erfordert engmaschige Überwachung und Überprüfung Nach Schätzungen des Bundesgesundheitsministeriums soll es ca. 400.000 verschiedene Medizinprodukte geben. Dazu zählen Geräte für (Labor-) Diagnostik und Intensivmedizin ebenso wie Implantate, Instrumente, Verbandmittel oder OP-Material. Je komplexer das Wirkprinzip und die Funktionsweise von Medizintechnikprodukten ist, desto genauer müssen diese Produkte entwickelt, deren Entwicklung dokumentiert, ihre Wirkweise geprüft, die Sicherheit gewährleistet und die Leistung im Markt beobachtet werden. Oberstes Prinzip bei der Zulassung von Medizintechnikprodukten ist das Gewährleisten der allzeitigen Sicherheit und eines beständig hohen Leistungsniveaus (Safety and Performance) des Produkts: Dazu gehören die Risikoanalyse und die Risikobewertung zum Nachweis der Sicherheit, die Durchführung einer klinischen Bewertung bzw. Prüfung zum Nachweis der Leistungsfähigkeit und Wirksamkeit, sowie ein umfassendes Qualitätsmanagementsystem: Medizinprodukte müssen nachweislich in der Lage sein, die in der Produktkennzeichnung beschriebene technische Leistung zu erbringen, um die vom Hersteller ausgelobte medizinische Funktion erfüllen zu können. Übrigens: Die Anforderungen an die Hersteller von Medizinprodukten sind von der Regelungsdichte vergleichbar mit denen bei Arzneimitteln.
Transparente Implantate Die Sicherheit eines Medizintechnikprodukts bezieht sich dabei auf alle einzelnen Komponenten, Bestandteile und Entwicklungsschritte des Produkts und beginnt mit der ersten Notiz in einem Entwicklungsdokument. Als Bestandteil eines Medizinprodukts gilt nicht nur das Material, aus dem es gefertigt wird, und die Geometrie, die das Produkt definiert: Ein Medizinprodukt definiert sich (mindestens) aus dem Material, der Geometrie, der Wirkweise, dem Herstellungsverfahren und der Veredelungsschritte. Für all diese Merkmale eines Medizintechnikprodukts gilt – für sich allein und in Kombination miteinander – der Anspruch auf Sicherheit und beständig hohes Leistungsniveau. Um „Safety and Performance“ eines Medizintechnikprodukts überprüfen, bewerten und voraussagen (künftig gewährleisten) zu können, ist es wichtig, dass alle Entwicklungsschritte bei der Fertigung einer Komponente eines Medizintechnikprodukts geplant, dokumentiert, jederzeit nachvollziehbar und mit beherrschbarem (Rest-) Risiko versehen umgesetzt werden.
Medical Device Directive, Medical Device Regulation und mitgeltende Normen Die Anforderungen an die Entwicklung eines Medizinprodukts sind klar definiert und müssen eingehalten werden. Folgt eine Entwicklung nicht den in der Medical Device Directive/Medical Device Regulation und den in den mitgeltenden Normen definierten Vorgehensweisen, so kann ein Produkt nicht auf den Markt gebracht werden. Folgt die Fertigstellung eines Medizintechnikprodukts nicht einer gelenkten Entwicklung, kann weder Sicherheit, noch ein gleichbleibendes hohes Leistungsniveau angenommen werden. Die Aufsichtsbehörden werden zurecht die Zulassung und Vermarktung verwehren.
Sackgassen in Entwicklung und regulatorischer Umsetzung frühzeitig erkennen Die allgemeine Gesetzesgrundlage für das „Inverkehrbringen von Medizinprodukten“ sowie die mitgeltenden Normen definieren die „Straßenverkehrsordnung“ der eingeschlagenen „Entwicklungs- und Fertigungsroute“. Um zu vermeiden, dass zeit- und kostenintensive Forschungs- und Entwicklungsarbeit am Ende in einer Sackgasse feststeckt, müssen bereits vor Beginn die einschlägigen Gesetzestexte und Normen konsultiert werden. Hier werden in der „Stunde Null“ der Produktentwicklung bereits die Weichen gestellt: Die ISO 13485:2016 beschreibt im Kapitel 7 detailliert die Schritte, die es während der Entwicklung einzuhalten gilt. Die ISO 14971:2012 gibt Anleitung, wie das Risiko eines Produkts oder dessen Einzelkomponenten analysiert, bewertet und beherrscht werden müssen. Die ISO 62366 schreibt Entwicklungsmethoden vor, die die Gebrauchstauglichkeit eines Produkts oder seiner Komponenten gewährleisten.
Biologische Beurteilung von Medizinprodukten Von besonderer Bedeutung für die biochemische Entwicklung/Veredelung von Medizinprodukten oder einzelner Komponenten ist die ISO-Normenreihe 10993 zur biologischen Beurteilung von Medizinprodukten. Ziel der Norm ist es, die biologische Beurteilung hinsichtlich der Verträglichkeit der eingesetzten Materialien in Interaktion mit dem Körper zu bewerten. Untersucht werden dabei nicht nur die fertigen Produkte, sondern ebenfalls (und verpflichtend) auch die einzelnen Ausgangsstoffe sowie deren Wirkweise (einzeln und in Kombination miteinander), die zur Herstellung von Medizinprodukten oder eines Bestandteils verwendet werden. Die Normenreihe 10993 beschränkt sich dabei nicht nur auf implantierbare Medizinprodukte, sondern betrifft – sofern anwendbar – jede Art von Medizinprodukten. Außer der biologischen Prüfung beinhaltet die Norm zusätzlich physikalisch-chemische Prüfungen und Analysen von gelösten Stoffen und Substanzen und schreibt das Einhalten von Grenzwerten bei herauslösbaren Substanzen vor.
Forschung und Entwicklung zum Wohle des Patienten Viele – an sich dem Patienten dienliche – Forschungsergebnisse können nicht in der Praxis angewandt werden, da bei Forschung und Entwicklung verpflichtende Arbeits-, Dokumentations-, Verifizierungs- und Validierungsverpflichtungen nicht beachtet werden. Der Weg zur Zertifizierung ist dann verbaut oder kann nur beschritten werden, indem aufwändige Tests und Dokumentationsverfahren wiederholt werden. Oftmals ist aber für diese Wiederholungsschleifen das Budget nicht vorhanden. Nur wer seinen Design- und Entwicklungsprozess von vorneherein normkonform und entsprechend der gesetzlichen Vorgaben plant, kann sich wiederholende „Ehrenrunden“ in der Entwicklung ausschließen und vermeiden, dass kosten- und zeitaufwändige Untersuchungen und Testverfahren wiederholt werden müssen, da Umsetzung und Dokumentation nicht den regulatorischen Vorgaben entsprechen. Nur wer in „Design und Development“ frühzeitig ein entsprechendes Qualitätsmanagementsystem implementiert, wird am Ende seine Forschungsergebnisse fruchtbringend für den Patienten und das Unternehmen auf den Markt bringen können.
Analysiert man den Markt der Implantate, so erkennt man schnell: Die Biochemie hat auf den meisten Implantatoberflächen bereits ihre Spuren hinterlassen. Seien es künstliche Hüften, Zahnimplantate, Wirbelsäulenimplantate oder Stents: Mit Hilfe von biochemischen Veredelungsmethoden wurden Implantate in den letzten 15 Jahren immer mehr zu einer Art Multitalent in den Händen der Ärzte und im Körper des Patienten: Dank biochemischer Beschichtungen wachsen Implantate schneller ein, werden von der Immunabwehr des Patienten nicht als Fremdkörper wahrgenommen, setzen Wirkstoffe genau dosiert und genau an der richtigen Stelle im Körper frei und wirken antibakteriell oder entzündungshemmend.
Komplexe Wirkweise erfordert engmaschige Überwachung und Überprüfung Nach Schätzungen des Bundesgesundheitsministeriums soll es ca. 400.000 verschiedene Medizinprodukte geben. Dazu zählen Geräte für (Labor-) Diagnostik und Intensivmedizin ebenso wie Implantate, Instrumente, Verbandmittel oder OP-Material. Je komplexer das Wirkprinzip und die Funktionsweise von Medizintechnikprodukten ist, desto genauer müssen diese Produkte entwickelt, deren Entwicklung dokumentiert, ihre Wirkweise geprüft, die Sicherheit gewährleistet und die Leistung im Markt beobachtet werden. Oberstes Prinzip bei der Zulassung von Medizintechnikprodukten ist das Gewährleisten der allzeitigen Sicherheit und eines beständig hohen Leistungsniveaus (Safety and Performance) des Produkts: Dazu gehören die Risikoanalyse und die Risikobewertung zum Nachweis der Sicherheit, die Durchführung einer klinischen Bewertung bzw. Prüfung zum Nachweis der Leistungsfähigkeit und Wirksamkeit, sowie ein umfassendes Qualitätsmanagementsystem: Medizinprodukte müssen nachweislich in der Lage sein, die in der Produktkennzeichnung beschriebene technische Leistung zu erbringen, um die vom Hersteller ausgelobte medizinische Funktion erfüllen zu können. Übrigens: Die Anforderungen an die Hersteller von Medizinprodukten sind von der Regelungsdichte vergleichbar mit denen bei Arzneimitteln.
Transparente Implantate Die Sicherheit eines Medizintechnikprodukts bezieht sich dabei auf alle einzelnen Komponenten, Bestandteile und Entwicklungsschritte des Produkts und beginnt mit der ersten Notiz in einem Entwicklungsdokument. Als Bestandteil eines Medizinprodukts gilt nicht nur das Material, aus dem es gefertigt wird, und die Geometrie, die das Produkt definiert: Ein Medizinprodukt definiert sich (mindestens) aus dem Material, der Geometrie, der Wirkweise, dem Herstellungsverfahren und der Veredelungsschritte. Für all diese Merkmale eines Medizintechnikprodukts gilt – für sich allein und in Kombination miteinander – der Anspruch auf Sicherheit und beständig hohes Leistungsniveau. Um „Safety and Performance“ eines Medizintechnikprodukts überprüfen, bewerten und voraussagen (künftig gewährleisten) zu können, ist es wichtig, dass alle Entwicklungsschritte bei der Fertigung einer Komponente eines Medizintechnikprodukts geplant, dokumentiert, jederzeit nachvollziehbar und mit beherrschbarem (Rest-) Risiko versehen umgesetzt werden.
Medical Device Directive, Medical Device Regulation und mitgeltende Normen Die Anforderungen an die Entwicklung eines Medizinprodukts sind klar definiert und müssen eingehalten werden. Folgt eine Entwicklung nicht den in der Medical Device Directive/Medical Device Regulation und den in den mitgeltenden Normen definierten Vorgehensweisen, so kann ein Produkt nicht auf den Markt gebracht werden. Folgt die Fertigstellung eines Medizintechnikprodukts nicht einer gelenkten Entwicklung, kann weder Sicherheit, noch ein gleichbleibendes hohes Leistungsniveau angenommen werden. Die Aufsichtsbehörden werden zurecht die Zulassung und Vermarktung verwehren.
Sackgassen in Entwicklung und regulatorischer Umsetzung frühzeitig erkennen Die allgemeine Gesetzesgrundlage für das „Inverkehrbringen von Medizinprodukten“ sowie die mitgeltenden Normen definieren die „Straßenverkehrsordnung“ der eingeschlagenen „Entwicklungs- und Fertigungsroute“. Um zu vermeiden, dass zeit- und kostenintensive Forschungs- und Entwicklungsarbeit am Ende in einer Sackgasse feststeckt, müssen bereits vor Beginn die einschlägigen Gesetzestexte und Normen konsultiert werden. Hier werden in der „Stunde Null“ der Produktentwicklung bereits die Weichen gestellt: Die ISO 13485:2016 beschreibt im Kapitel 7 detailliert die Schritte, die es während der Entwicklung einzuhalten gilt. Die ISO 14971:2012 gibt Anleitung, wie das Risiko eines Produkts oder dessen Einzelkomponenten analysiert, bewertet und beherrscht werden müssen. Die ISO 62366 schreibt Entwicklungsmethoden vor, die die Gebrauchstauglichkeit eines Produkts oder seiner Komponenten gewährleisten.
Biologische Beurteilung von Medizinprodukten Von besonderer Bedeutung für die biochemische Entwicklung/Veredelung von Medizinprodukten oder einzelner Komponenten ist die ISO-Normenreihe 10993 zur biologischen Beurteilung von Medizinprodukten. Ziel der Norm ist es, die biologische Beurteilung hinsichtlich der Verträglichkeit der eingesetzten Materialien in Interaktion mit dem Körper zu bewerten. Untersucht werden dabei nicht nur die fertigen Produkte, sondern ebenfalls (und verpflichtend) auch die einzelnen Ausgangsstoffe sowie deren Wirkweise (einzeln und in Kombination miteinander), die zur Herstellung von Medizinprodukten oder eines Bestandteils verwendet werden. Die Normenreihe 10993 beschränkt sich dabei nicht nur auf implantierbare Medizinprodukte, sondern betrifft – sofern anwendbar – jede Art von Medizinprodukten. Außer der biologischen Prüfung beinhaltet die Norm zusätzlich physikalisch-chemische Prüfungen und Analysen von gelösten Stoffen und Substanzen und schreibt das Einhalten von Grenzwerten bei herauslösbaren Substanzen vor.
Forschung und Entwicklung zum Wohle des Patienten Viele – an sich dem Patienten dienliche – Forschungsergebnisse können nicht in der Praxis angewandt werden, da bei Forschung und Entwicklung verpflichtende Arbeits-, Dokumentations-, Verifizierungs- und Validierungsverpflichtungen nicht beachtet werden. Der Weg zur Zertifizierung ist dann verbaut oder kann nur beschritten werden, indem aufwändige Tests und Dokumentationsverfahren wiederholt werden. Oftmals ist aber für diese Wiederholungsschleifen das Budget nicht vorhanden. Nur wer seinen Design- und Entwicklungsprozess von vorneherein normkonform und entsprechend der gesetzlichen Vorgaben plant, kann sich wiederholende „Ehrenrunden“ in der Entwicklung ausschließen und vermeiden, dass kosten- und zeitaufwändige Untersuchungen und Testverfahren wiederholt werden müssen, da Umsetzung und Dokumentation nicht den regulatorischen Vorgaben entsprechen. Nur wer in „Design und Development“ frühzeitig ein entsprechendes Qualitätsmanagementsystem implementiert, wird am Ende seine Forschungsergebnisse fruchtbringend für den Patienten und das Unternehmen auf den Markt bringen können.
ZUR PERSON
Dietmar Schaffarczyk ist Lehrbeauftragter an der Universität Tübingen und externer Auditor für Medizintechnik (Diplom SAQ). Zudem ist er als Quality-Experte von der European Organization for Quality zertifiziert und unterstützt Unternehmen bei Fragen zur Entwicklung und Zulassung von Medizinprodukten. Außerdem führt Schaffarczyk die Geschäfte von Stimos, einem Unternehmen, das sich auf die (bio)chemische Funktionalisierung von Implantatoberflächen spezialisiert hat.
Kontakt
Stimos GmbH
Byk-Gulden-Sraße 2 / F21
78467 Konstanz
Deutschland









