
Trends in der Anlagenqualifizierung
Neue Wege der pharmazeutischen Industrie
-
 © Eisenhans | Fotolia.com
© Eisenhans | Fotolia.com -
 Dipl.-Ing. R. Gengenbach, Gempex
Dipl.-Ing. R. Gengenbach, Gempex -
 Abb. 1: Regulatorische Entwicklung Qualifizierung
Abb. 1: Regulatorische Entwicklung Qualifizierung -
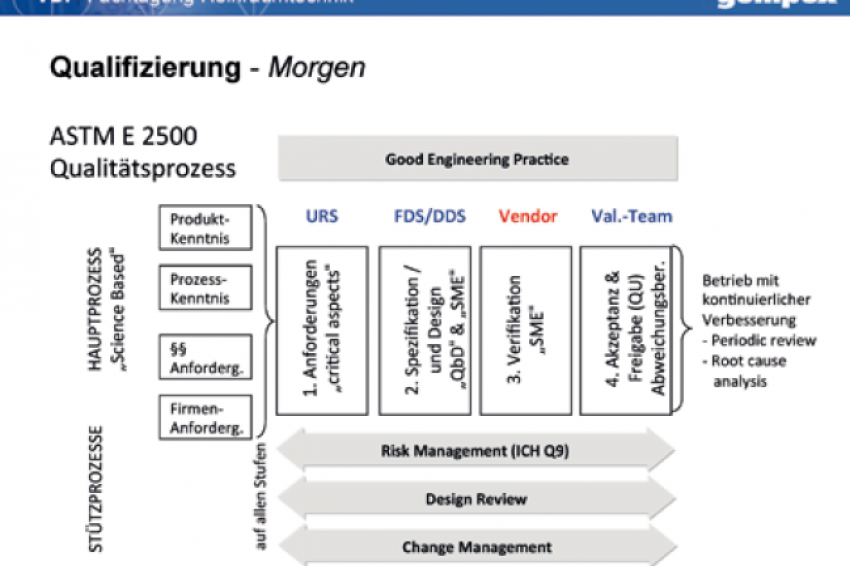 Abb. 2: Ablauf Qualifizierung nach ASTM E2500
Abb. 2: Ablauf Qualifizierung nach ASTM E2500
Die Hintergründe Qualifizierung ist die dokumentierte Beweisführung, dass ein Technisches System ordnungsgemäß, den Wünschen entsprechend konstruiert und installiert ist und dass es genau so funktioniert wie ursprünglich vom Nutzer spezifiziert. Qualifizierung ist ein Teil der Validierung, die neben der Technik zusätzlich die Prozesse und Verfahren bei der Beweisführung berücksichtigt. Beide Elemente, Qualifizierung der Technik und die Validierung von Prozessen und Verfahren sind über die Regeln der Guten Herstellungspraxis (engl. GMP = Good Manufacturing Practices) insbesondere in der pharmazeutischen Industrie verbindlich gefordert und fester Bestandteil bei behördlichen Inspektionen.
Eine erste Richtlinie zur Praxis der Validierung (einschließlich Qualifizierung) wurde 1983 von der FIP (Fédération International de Pharmaceutique) [1] herausgegeben mit ersten Hinweisen und Empfehlungen zur konkreten Durchführung. Seitdem sind nunmehr 30 Jahre vergangen, in denen sich das Thema weiter entwickelt und weiter detailliert hat. Ein Blick auf den Zeitstrahl (Abb. 1) zeigt dabei schnell, wie sich nicht nur das Thema, sondern auch die zugehörigen Regelwerke und Richtlinien nahezu exponentiell entwickelt haben. Gab es in den Anfängen etwa alle drei bis vier Jahre eine regulatorische Neuerung, so steigerte sich dies auf drei bis vier Neuerungen und damit verbundenen neuen Richtlinien und Regelwerke, die seit 2.000 etwa jährlich erscheinen. Eine Frequenz, mit der nur noch schwer Schritt gehalten werden kann, zumal die Herausgabe neuer Richtlinien auch mit neuen Anforderungen an die inhaltliche Bearbeitung des Themas verbunden ist. Aber nicht nur die Fülle der Regelwerke ist bei dem Thema „Qualifizierung" eine Herausforderung, auch die Fülle des im Rahmen der Qualifizierung entstehenden Papiers (Prüf-, Testpläne und -berichte).
Kein Wunder also, dass man sich schon früh damit beschäftigte, wie man diese Papierflut bewältigen, besser noch reduzieren kann. Konzentrierten sich die ersten Ansätze auf das Thema „Reduktion durch Selektion", d.h., Qualifizierung nur noch von ausgewählten kritischen technischen Systemen (z.B. gezielte Auswahl mit Hilfe von Risikoanalysen), so geht heute der Trend verstärkt hin zur „Reduktion durch Selektion plus Integration".
Das bedeutet, man möchte Aufwand und Papier nicht nur durch gezielte Selektion kritischer technischer Systeme reduzieren, man möchte auch so viel als möglich von ingenieurtechnischer Seite bereits durchge führte Prüfungen und Tests in der Qualifizierung mit berücksichtigen (integrieren) und auf diese Weise Doppelarbeit sparen. Die nachfolgenden Ausführungen beleuchten die Qualifizierung „Gestern - Heute - Morgen" und versuchen darzulegen, wo die Zukunft bei der modernen Qualifizierung liegen könnte und welche Herausforderungen dies an die Technik, insbesondere an die Zulieferer stellt.
Qualifizierung "Gestern"
Im Rahmen der Qualifizierung wurden schon früh die heute allseits bekannten Elemente der Installationsqualifizierung (IQ = Installation Qualification), der Funktionsqualifizierung (OQ = Operational Qualification) und der Leistungsqualifizierung (PQ = Performance Qualification) unterschieden. Im Rahmen der IQ wird die korrekte Spezifikation und Installation, im Rahmen der OQ die korrekte Funktion und im Rahmen der PQ die Leistungsfähigkeit des entsprechenden technischen Systems nachgewiesen und dokumentiert.
Dabei war es von jeher eine Grundvoraussetzung, dass diese Beweisführung geplant und schriftlich erfolgt. Auf der Dokumentenseite wurde auch schon früh ein Masterplan (VMP = Validation Master Plan) gefordert, in dem das Gesamtprojekt skizziert und die notwendigen Einzelaktionen einschließlich erforderlicher Ressourcen aufgeführt sein müssen. Für die Einzelaktionen (IQ, OQ, PQ) wurden und werden noch heute Pläne erwartet, in denen die beabsichtigte Vorgehensweise, die Details und insbesondere die Akzeptanzkriterien beschrieben werden. Die Pläne einschließlich aller zugehöriger Checklisten und Rohprüfdokumente sind durch das Validierungsteam - ein Team interdisziplinärer Fachexperten - zu prüfen und durch die Qualitätseinheit des Systemeigners formal mit Unterschrift zur Durchführung freizugeben.
Nach Durchführung erfolgt ein ähnliches Prozedere für die Qualifizierungsergebnisse und den resultierenden Bericht. Auch dies wird inhaltlich durch das Validierungsteam geprüft und letztendlich durch die Qualitätseinheit formal freigegeben. Für die Durchführung selbst wurden nicht selten sehr detaillierte und systemspezifische Checklisten entwickelt, in die, ausgehend von der technischen Dokumentation, Spezifikations- und Funktionskriterien mit Sollwertvorgaben als Prüfgrundlage eingetragen wurden. Und nicht selten wurden die Details von der Anlage selbst oder von den technischen Unterlagen in die Checklisten übernommen um dann umgekehrt zu bestätigen, dass sich diese Details vor Ort und in der technischen Dokumentation auch wirklich finden.
Insbesondere die in USA entwickelten Systeme zeigten diese Papier generierende Symptomatik. Hatte man ein Projekt, welches zum Beispiel nur 10 technische Komponenten umfasste, so hatte man alleine hierfür - ohne Masterdokumente - für die Elemente IQ, OQ und PQ insgesamt 30 Dokumente mit allen zugehörigen Checklisten und Anhängen zu erzeugen. Und Projekte umfassen üblicherweise deutlich mehr als nur 10 Komponenten.
Qualifizierung "Heute"
Ende der 90er Jahre und mit Beginn des 21. Jahrhunderts wurde die Industrie selbstkritischer und hat erkannt, dass die stur formalistische Vorgehensweise zwar jede Menge Papier und Kosten erzeugt, die Qualität aber nicht in gleichem Ausmaß verbessert. Ganz im Gegenteil, die Menge an Papier und der strenge Formalismus haben oft wirklich kritische Probleme erst gar nicht erkennen lassen. Entsprechend hat sich die Philosophie der Industrie aber auch der Behörde heute deutlich geändert. Zum einen hat man ein weiteres Element, die Design Qualifizierung (DQ = Design Qualification), in die Aktivitäten mit aufgenommen, weil man erkannt hat, dass die meisten Fehler schon in der Planung gemacht und daher genau dort ausgeschlossen werden müssen.
Zum anderen hat man das Element Risikoanalyse als das wichtigste „GMP Tool" eingeführt um u.a. zu selektieren, welche technischen Komponenten denn kritisch sind und wirklich qualifiziert werden müssen und welche eher unkritisch sind, und daher keine formale Qualifizierung benötigen. Man hat weiter erkannt, dass es nicht zwingend notwendig und nicht unbedingt hilfreich ist, wenn man alle Nachweisprüfungen immer in detaillierten Checklisten abbildet. Vielmehr macht es Sinn, direkt technische Unterlagen (z.B. Rohrleitungs- und Instrumentenfließbilder, Konstruktionszeichnungen, Elektropläne, Stücklisten) als Prüfgrundlage zu nutzen und auch bereits durchgeführte Prüfungen der Ingenieure aus den Hersteller- und Abnahmeprüfungen (FAT = Factory Acceptance Tests, SAT = Site Acceptance Tests) mit in die Qualifizierung einzubinden. Trotz all dieser Maßnahmen wird die Vorgehensweise bei der Qualifizierung heute noch immer nicht als optimal und zielführend empfunden.
Die Designqualifizierung ist in ihrem Grundverständnis zwar klar, die Vorgehensweise aber nicht. Jede Firma führt die DQ anders aus. Während die einen sehr detailliert und systematisch Punkt für Punkt die Betreiberanforderungen (Lastenheft) den Ausführungsempfehlungen des Herstellers (Pflichtenheft) gegenüber stellen, verstehen andere unter der DQ die reine Prüfung von Ausführungszeichnungen oder auch nur das Erstellen eines Lastenheftes. Die Risikoanalysen zur Identifizierung kritischer, qualifizierungsrelevanter technischer Systeme werden häufig sehr formalistisch und mehr zum Selbstzweck als zur wirklichen Aufwandsreduzierung durchgeführt. Auch bei den Qualifizierungsplänen findet sich noch keine einheitliche Praxis.
Während die einen den Umfang schon mutig auf das Notwendige reduziert haben, folgen andere noch immer dem Checklistenprinzip. Und schließlich die Einbindung von FAT und SAT Ergebnissen - hier hat man schnell die Erkenntnis gewonnen, dass dies natürlich nur funktionieren kann, wenn es eine gute Ingenieurspraxis (GEP = Good Engineering Practice) mit entspre chend ordentlich durchgeführten und dokumentierten Prüfungen gibt. Und gerade das ist leider noch immer selten der Fall.
In ihrem „White Paper" [2] vom März 2005 macht die ISPE (International Society for Pharmaceutical Engineering) diese Problematik deutlich. Ein eigens ins Leben gerufenes „Qualification Task Team" hält unverblümt fest, dass es aus Sicht der Experten derzeit kein wirklich effizientes und wirklich wirksames Qualifizierungssystem gibt. Dass die Systeme und Vorgehensweisen noch immer zu formalistisch, zu aufwändig und zu teuer sind und nicht ausreichend auf die Patientensicherheit fokussieren. Es wird zum Ausdruck gebracht, dass es dringend an der Zeit sei, ein angemessenes modernes Qualifizierungskonzept zu entwickeln.
Qualifizierung „Morgen"
Das ISPE Qualification Task Team legt ein 10 Punkte Programm auf, dessen Forderungen durch weiterentwickelte Standards und Normen umgesetzt werden sollen. Kernpunkte sind nach wie vor ein risikobasiertes Vorgehen, die Einbindung der Herstellertests (FAT und SAT) sowie pragmatische und praxisorientierte Qualifizierungsdokumente. Die Forderungen der ISPE gehen jedoch noch weiter und sehen beispielsweise die Möglichkeit, bei Standardausrüstung die Qualifizierung drastisch zu reduzieren, gegebenenfalls durch eine Lieferantenqualifizierung zu ersetzen.
Generell möchte man gerade die IQ und OQ Aktivitäten stark zurücknehmen und hier vielmehr auf die Herstellertests verweisen. Man sieht dies als Hauptaufgabe der Technik und nicht als Hauptaufgabe einer pharmazeutischen Qualitätseinheit. Der Schwerpunkt des Nutzers sollte eindeutig auf der PQ, der Leistungsprüfung des technischen Systems liegen. Eine Norm, die genau dieses Ziel verfolgt, ist der 2007 von der ASTM herausgegebene Standard ASTM E2500 [3]. Er beschäftigt sich mit Gebäude-, Prozess- und Hilfseinrichtungen sowie mit Prozessüberwachungs-, Regelungs- und Automatisierungseinrichtungen, die allgemein unter dem Begriff „Herstelleinrichtungen" (Manufacturing Systems) zusammengefasst werden.
Eine vergleichbare Norm, der Standard ASTM E2537 [4], ist im Februar 2008 zum Thema „Herstellung" (Manufacturing) erschienen. Beide Normen nutzen neu den Überbegriff „Verifizierung" (Verification) und fassen darunter sowohl die „üblichen" technischen Standardprüfungen als auch die formalen Qualifizierungs- und Validierungsaktivitäten zusammen. Im ASTM E2500 Standard werden die oben bereits beschriebenen Themen „risikobasiertes Vorgehen" und „Nutzung von Herstellerunterlagen bzw. -tests" als Schlüsselelemente herausgestellt. Es wird aber weiter auch auf „wissenschaftsbasierte Vorgehensweisen", auf „kritische Aspekte" der Herstellsysteme, auf den „Fachexperten" (Subject Matter Expert) und auf eine kontinuierliche Prozessverbesserung hingewiesen. Kernstück des normativen Leitfadens stellt ein Ablaufschema dar, das in vereinfachter Ausführung in Abbildung 2 wiedergegeben wird.
Bei diesem „idealisierten" Ablauf geht man von Haupt- und Stützprozessen aus. Bei den Hauptprozessen werden unter Zugrundelegung von Produkt- und Prozesskenntnissen sowie unter Berücksichtigung regulatorischer und firmeninterner Vorgaben die generellen Anforderungen und die „critical aspects" definiert und in einem Lastenheft (engl. URS = User Requirement Specification) beschrieben. Die Fachexperten entwickeln weiter die funktionalen und Detail Spezifikationen (FDS = Functional Design Specification, DDS = Detail Design Specification) und achten hier bereits auf alle relevanten Qualitätsanforderungen (QbD = Quality by Design). Nach der Realisierungsphase schließt sich dann die bekannte Test- und Qualifizierungsphase an, die hier jetzt unter dem Begriff „Verification" zusammengefasst und in die Obhut des „Subject Matter Experts", also dem Fachexperten (auch Hersteller oder Lieferant) gegeben wird.
Erst danach kommt die Qualitätseinheit ins Spiel, die erst ganz am Ende die Verifikationsergebnisse formal abnimmt und das technische System freigibt, ggf. begleitet von einem Abweichungsbericht. Begleitet wird dieser Ablauf durch Risikoanalysen, durch Design Review Aktivitäten und durch ein Change Management, die als Stützprozesse bei allen Schritten zum tragen kommen und die überdies eine Gute Ingenieurspraxis voraussetzen.
Qualifizierung "Übermorgen"
Auf den ersten Blick scheint dieser normative Vorschlag ziemlich deckungsgleich mit den bisherigen, bereits etablierten Abläufen zu sein. Erst beim mehrmaligen Lesen und bei Konzentration auf die Details erkennt man jedoch die eigentliche Absicht und die geplanten Prozessverbesserungen. Ein Schwerpunkt liegt ganz sicher in der Identifizierung der „critical aspects", die hier bewusst nicht auf die „quality critrical aspects" reduziert wurden. Vielmehr geht es darum, unter dem Aspekt einer „Good Engineering Practice" tatsächlich von Anfang an alle wesentlichen und kritischen Eigenschaften und Elemente des technischen Systems zu bestimmen und beim Design und der Ausführung auf diese Punkte zu achten, was ein entsprechendes Qualitätsbewusstsein und Qualitätssystem beim Hersteller voraussetzt.
Ein weiterer Schwerpunkt liegt bei diesem Konzept in der Zuordnung der „Verifikationsaktiviäten" zu den Fachexperten, d. h., diejenigen, die das technische System am besten kennen, sollen dieses am Ende auch auf Herz und Nieren prüfen und die Tauglichkeit bestätigen. Im Prinzip ein Vorgehen, wie es heute schon unterschwellig stattfindet. Nicht selten ist es der Hersteller oder Lieferant, der die wirklich kritischen und relevanten Tests durchführt und die danach in der Qualifizierung wiederholt werden, um dem formale System zu genügen.
Im Falle eines Vorgehens nach ASTM E2500 wäre dies dann lediglich „offiziell". Schließlich stellt das Konzept dann noch klar heraus, dass das Thema Risikoanalyse keine einmalige Angelegenheit, sondern ein kontinuierlicher Prozess ist, der auf mehreren Projektstufen gestützt durch ein kontinuierliches Design Review stattfindet und der natürlich stets ein „Good Engineering Practice" voraussetzt. Das Zukunftskonzept könnte also so aussehen, dass man sich geeignete und qualifizierte Lieferanten sucht, die mit dem Thema Qualität und Qualifizierung bestens vertraut sind und daher von Anfang an die gesamten Elemente IQ und OQ vollumfänglich abdecken und nach ASTM E2500 oder einer vergleichbaren Norm arbeiten. Ein einfacher Download des zugehörigen Zertifikates aus dem Internet wäre der letzte verbleibende formale Akt. Die PQ, als harter Leistungstest wäre das, was auch weiterhin durch den Betreiber geleistet werden müsste, was sicher aber auch in dessen Kompetenzbereich liegt.
Die Herausforderung
Mit den von der ISPE im White Paper getätigten Äußerungen ist sicher ein reales und weithin erkanntes Problem angesprochen. Und die in den zitierten ASTM Standards beschriebenen Zukunftskonzepte scheinen vernünftig und durchaus realitätsnah zu sein. Alleine die Tatsache, dass die Diskussion dieser Probleme bereits wieder acht Jahre zurückreicht und eine Umsetzung - zumindest hier in Europa - nicht wirklich erkennbar ist zeigt, dass die eigentlichen Probleme tiefer liegen müssen.
Sie liegen noch immer - betrachtet man sich die Praxis - in der nicht wirklich existenten „Guten Ingenieurspraxis", die ein Thema wie die Qualifizierung vollumfänglich integrieren würde. Sie liegen in fehlenden Leitlinien für die Hersteller, die beschreiben, wie diese eine Qualifizierung oder auch nur eine formale Verifizierung anforderungsgerecht durchführen sollten. Und sie liegen im noch immer konservativen Denken der pharmazeutischen Industrie und der Behörden, die an dieser Stelle - vielleicht auch zurecht - nicht bereit sind, einen wichtigen Teil der Qualitätssicherung aus den Händen zu geben und an die Hersteller zu übertragen. Es bleibt daher nach 30 Jahren Qualifizierung und nach mindestens acht Jahren Optimierungsbemühungen noch immer eine große Herausforderung, dieses Thema dahingehend in den Griff zu bekommen, dass die Konzepte als effizient, wirksam und wirtschaftlich bezeichnet werden können.
Literaturverzeichnis auf Anfrage vom Autor erhältlich.
Kontakt
Dipl.-Ing.R.Gengenbach
Gempex GmbH, Mannheim
Tel.: +49 621 819119 0
r-gengenbach@gempex.com
Kontakt
gempex GmbH – THE GMP-EXPERT
Besselstraße 6
68219 Mannheim
Deutschland
+49 621 819119 0
+49 621 819119 40









